
Getting to the Bottom of My Health: Biotin and (V)LCAD
My leading hypothesis after six months of research, testing, and self-experiments on the path to biochemical optimization.

For nearly 20 years, I have strongly suspected that I have a genetic disorder in synthesizing or recycling something that is absent or poorly available from plant foods, is very rich in organ meats, and is not present in any of the supplements I was taking when I was a vegan.
After six months of research, testing, and self-experimenting, I now believe what I have been looking for is a mix of moderate genetic defects in biotin recycling, possibly combined with poor cellular uptake of biotin, and definitely combined with a defect in burning long-chain fatty acids for energy. The treatments for this, which I will be testing one at a time, will be high-dose biotin, high-dose riboflavin, possibly high-dose L-carnitine and glycine, and, if needed, an otherwise low-fat diet supplemented with C8-specific MCT oil.
Let’s step all the way back to the beginning of my first suspicion as I walk you through how I have arrived at the new hypothesis.
The Origin of the Genetic Defect Suspicion
The basis for the original suspicion is as follows.
I went vegetarian, and six months later vegan, beginning at some point circa 1999 when I was 18 years old, and this led my entire body, but especially my teeth and my mind, to fall apart. While I sought no diagnosis, I consider myself to have gone psychotic or something near to it at that point, as my paranoia had made daily life so difficult that I was often afraid to so much as eat. I also believe that this is one of two times in my life where I am missing many memories, the other being when I smoked large amounts of marijuana from ages 14-15.
I first wrote about this in 2005, and I updated my reflections on it in 2013.
I was vegan for a total of a year, sandwiched inside two six-month periods of less strict vegetarianism on either end, for a total of two years avoiding animal foods.
When I discovered the work of Weston Price and began eating at least three egg yolks per day, a pound of buffalo liver per week, and otherwise focusing on omnivorous nutrient-density, these problems completely disappeared.
While I do not believe strict veganism without any shellfish can ever be a robustly nutritionally optimized diet, it is also the case that few people have such night-and-day experiences of neurological catastrophe and salvation between veganism and omnivory as I had. I know enough ex-vegans to know that my experience is much more extreme than is typical.
This suggests that something sets me apart in having a much more extreme intolerance of animal food deprivation than most people have, something presumably inborn, rooted in my genetics.
On the one hand, there are many nutritional problems that can arise on a plant-only diet and it is possible that I have a collision of many small genetic variants that make me mediocre on each of these and thus very broadly intolerant of veganism. On the other hand, the idea that there could be a very simple explanation with one genetic defect to rule them all has always been appealing for its simplicity. Were it true, discovering it would probably lead to my greatest personal biohack of all time.
Other Health Problems in the Life After Veganism
Despite the night-and-day reversal of the major health problems I had when I was vegan, my post-Weston Price health was not perfect.
A Subtle Problem of Fat Intolerance
One subtle point I realized was that when I ate very high-fat, I felt fatigued, foggy-headed in the afternoon, and felt like I was developing a lump in my throat. Moderating my fat intake seemed to help with this.
A Serious Problem of Fasting Intolerance
A much stronger problem I had was fasting intolerance. As an Orthodox Christian, I would fast before communion on Sundays. I always arrived early to help chant the Orthros, the service before the Divine Liturgy (analogous to what Catholics call Matins and Mass), but I would often briefly pass out while standing and chanting, awakened suddenly by nearly falling over. A small amount of juice or sugar offered enormous relief, suggesting I was getting hypoglycemic. It is difficult to remember for sure, but I believe this was much worse while vegan, but still a problem afterward.
Another manifestation of this fasting intolerance was that I would wake up repeatedly during the night and need to eat something to fall back asleep, even if I didn’t feel hungry.
My late friend Paul Idol (rest in peace) had recommended I read Mastering Leptin by Byron Richards. This book had recommendations such as eating no more than three and preferably only two times a day, fasting for 5-6 hours between each meal and 11-13 hours overnight, and fasting for 3 hours before exercise. I was completely unable to follow these, so Paul recommended I supplement with acetyl-L-carnitine and R-lipoic acid. I took one capsule of each of these at each meal. I do not remember the doses, but from perusing supplements currently available online it looks like I was most likely getting daily totals of 1500 milligrams of acetyl-L-carnitine and 300 milligrams of R-lipoic acid. The R-lipoic acid supplements always contained D-biotin, and I was likely getting 450 micrograms per day of that as well.
This cocktail allowed me to put the Mastering Leptin recommendations in place. I do not remember how long I took it for, but I do remember that for years whenever I would feel like it was getting hard to go without meals, I would go back to it.
Much later, in grad school, I took up the practice of taking no food until the evening, when I would eat one vegan meal, on Wednesdays, Fridays, and during the weekdays of Lent and other fasting periods on the Orthodox calendar. I felt great doing this as long as I included coconut oil in these meals (which typically were thick lentil soup with sourdough whole grain toast), and as long as on non-fasting days I minimized my muscle meat consumption and ate the vast majority of my meat in the form of liver and heart. My heart-to-liver ratio was generally between 3:1 and 4:1.
An interesting parallel between this diet and the supplement cocktail is that liver, kidney, and heart are the richest sources of lipoic acid, with heart being about 2 to 6 times richer than liver. Liver is also the best source of biotin besides egg yolk. One discrepancy, however, is that skeletal muscle seems to be at least as rich in total carnitine as heart muscle, although heart is by far and away the best food source of coenzyme Q10.
Thus, the main commonalities between the supplement cocktail and dietary regime were lipoic acid and biotin.
Nevertheless, the cumulative stress and sleep deprivation of grad school made my fasting tolerance deteriorate over time. After that, I spent many years focusing on eating regularly because it made me feel better. It was my impression after all this time that nothing improved my fasting tolerance (as in my ability to skip breakfast or go a long time between meals) more than eating and sleeping. However, I never made a serious attempt to retest the supplement cocktail or the mostly-liver-and-heart routine.
Over the last year, I have gravitated toward fasting through the course of my morning routine, ending with a cold shower before breakfast, and have seemed to tolerate this well.
Vulnerability to Eczema and Cutaneous Fungal Infections
Winding back the clock to the early days post-vegan, another problem I developed is terrible, severe, whole-body eczema. What made this disappear was either the soil-based probiotic Primal Defense, or the Green Pasture X-Factor Butter Oil, or some combination of the two. I started the Primal Defense two weeks before I started the butter oil. The eczema was hugely improved about one month after adding the Primal Defense, and about two weeks after adding the butter oil, and was gone within a few months.
After that, the eczema would occasionally come back on my wrists, but never threaten to take over my body. Years later, this transitioned from my wrists to my fingers.
While it seemed that stress, sleep, and gut health had some vague relation to it, for years it never posed a major threat, and there was no obvious curative effect of any probiotics I tried. Primal Defense had reformulated to remove Saccharomyces boulardii and include that only in Primal Defense Ultra. While I felt that Primal Defense Ultra was better for my gut health, even that had no obvious curative effect on the residual eczema.
My bias was to believe that the butter oil was probably not important, since I consumed a lot of butter, cream, and whole milk anyway. Because of this bias, I never stuck with the butter oil and never tested whether it could impact the residual eczema.
I never had a whole-body eczema attack again until 2017, when I had an indoor black mold problem and developed a toxic overload of barium that I traced to makeup I was using to shoot my videos and paint dust an air conditioner was blowing over me from a chipping windowsill while I slept. At this point, the skin rash seemed to be voraciously eating me alive, and a dermatologist told me it looked obviously fungal. I went on terbinafine, an anti-fungal, which dimmed the voraciousness of the attack enough for my immune system to start handling it. At first, every supplement I tried seemed dangerous, whether it was probiotic, prebiotic, a nutrient, oral, or topical, and I got scared to try anything after several times of seeming to wipe away progress the terbinafine had made. However, once the terbinafine really started getting it under control, I found what made it finally go away was increasing my vitamin A intake, using a half a shot of vodka per day to increase the activation of the vitamin A, and using tanning beds.
Looking back on my first attack of whole-body eczema, I’m sure it too was fungal, as there was not much difference in the appearance.
After this, I had the eczema/fungus/whatever return to devour my hands each winter until I instituted a very rigorous UV light exposure plan: when the UV index is consistently above 5, I get 40 minutes of unprotected sunshine per day around solar noon. When it is consistently below 5, I go tanning twice a week using beds that have a mix of UVA and UVB. Since I have instituted this and executed it systematically, I have had no problem with recurring eczema.
Vulnerability to Hypocalcemic Tetany
In the early aftermath of the first whole-body eczema episode, I tried various restrictive diets to try to find a way to get rid of the residual eczema that would often creep back onto my wrists.
One of those was a gluten-free, casein-free diet.
Once I cut out milk, I started suffering from convulsions while driving to church in the fasting state.
A doctor told me this was “anxiety,” but he seemed oblivious to the fact that anyone who started suffering from convulsions would of course be anxious about it.
I cured this as soon as I started supplementing bone meal, so it was an obvious case of hypocalcemic tetany.
Vulnerability to Assorted Neurological Problems
The night I began terbinafine in 2017, I developed the worst twitching problem of my life, with the large muscles of my thighs making big repeated jerks. I tested my response to all the electrolytes and found that none of them did anything except potassium. Quickly, I found that this problem 80% disappeared if got my potassium intake up to 5 grams per day. The remainder disappeared if I also avoided acidic foods and beverages.
Nevertheless, I remained vulnerable to small slips in my diet causing my eyes to twitch. Even without slips, working out would cause the exercised muscles to start twitching, I had terrible post-workout fatigue that would lay me out for five days. Even one sip of wine would start a twitching cascade, especially if I hadn’t eaten yet.
As I described in How Measuring My Urine pH Got Me to Love Working Out Again, I fixed these vulnerabilities with baking soda. As I described in Three Health Issues Converge on One Bone, the twitching receded into a light paresthesia on my left cheek, which became more and more subtle over time until it mostly disappeared.
Nevertheless, I was worried about long-term effects of baking soda impairing my digestion, and I also found that overdosing on it could cause a different kind of twitching fixed by calcium and acidic foods. This is because alkalinity makes calcium ions in the blood bind to the protein albumin and become biologically unavailable.
I ultimately developed a simple solution to this: 1/4 to 1/2 teaspoon a day of beta-alanine, and 25 milligrams of vitamin B6 in the form of pyridoxal 5’-phosphate (P5P), per day. The beta-alanine is used to synthesize carnosine, which buffers acidity inside muscles before it spills into the circulation. Acidity leads to the hydrolysis of glutamine to form glutamate, which can cause excessive neuronal stimulation. P5P helps get rid of the glutamate by converting it to GABA, which has the opposite neuronal effects, or to alpha-ketoglutarate so it can be burned for energy in the citric acid cycle.
Numerous times I tried stopping this cocktail, and every time the twitching would start to come back within two weeks. Then, at the beginning of 2022, I eliminated it for good and the twitching never came back. This indicates to me that terbinafine caused mitochondrial damage that took 4.5 years to heal, but while it was healing my supplement cocktail rendered it completely asymptomatic.
A second problem that arose when I was taking the terbinafine was a gait abnormality where my foot seemed to drag on the ground unless I spent excessive effort lifting it into the air. I am not sure what made it go away, but it might have spontaneously disappeared when I went off the terbinafine after six weeks.
This same gait abnormality reared its head again in 2019. I had been playing around with B vitamins in my diet and had recently added 500 milligrams per day of pantothenic acid. Since pantothenic acid shares an intestinal transporter with lipoic acid and biotin, I added lipoic acid and biotin to my protocol, taking each of the three supplements at a different meal. This appeared to cure it entirely.
I went to a neurologist to check it out, and ran lots of tests that found nothing. However, this may be because I cured myself by balancing B vitamins before I ever went to the neurologist. Garrett Smith thought I was suffering from vitamin A toxicity, and I wrote up a detailed response describing all of this.
This vulnerability to intermittent neurological problems bothers me because it suggests something is still a little off under the hood that could go terribly wrong with a sufficient insult to my system. For example, what if I get another fungal infection and am driven to dose myself with another round of mitochondrial damage to get rid of it? How many times can I do this and bounce back better than before?
We can now continue this concern by fast forwarding to the turn of 2022, with the series of events that drove me to become serious about biochemical optimization.
The Lead-In to My 2022 Goal of Biochemical Optimization
During Lent of 2022, I temporarily changed my diet along the lines with what I advised in How to Eat Well During an Orthodox Lent. The main changes were that I removed eggs and the Ancestral supplement I was taking to provide the equivalent of one ounce of liver per day, I replaced dairy products with a calcium supplement, and I replaced my meat with lentils and a rotation of shellfish.
Anxiety From Not Walking and Under-Eating
In parallel with this, I had a completely independent source of anxiety arise. During physical therapy, we tried cupping my calves. While this proved to provide an incredible increase in ankle mobility, it was extremely painful. I noticed ropy structures in my calves, and initially I, my PT, and the Functional Patterns trainer I was working with all thought the solution was more manual therapy. However, I noticed that through all of this painful therapy that the ropy structures were only getting worse. My PT then thought they might be varicose veins that were brought up to the surface through irritation, and we decided to rest my calves, which in fact made the ropy structures go away.
To fully rest my calves, I eliminated walking for a few weeks. Ordinarily between my morning walk and working at my treadmill desk I would get 1-2 hours of walking per day.
At this point I started feeling a physiological wave of anxiety overwhelm me in the late afternoon.
With time, I realized that I had been preventing this anxiety with walking, and that it was highly correlated with under-eating when I was distracted by work.
Even though the anxiety was physiologically driven, it impacted my psychology. For example, even though I knew it was provoked by under-eating, it would make me irrationally afraid that it would get worse upon eating. At first I solved this by dissipating the anxiety through walking on my treadmill, and then eating once the anxiety was gone.
However, I eventually settled into a very proactive regime involving a morning walk and some treadmill time, as well as always eating breakfast and lunch before the late afternoon. By being very proactive about both walking and eating, the anxiety went away.
A Neurological or Muscular Problem During Lent
Nevertheless, this weeks-long process coincided with developing what I believe to be a very real neurological or muscular problem that was caused by my Lenten diet.
As Lent progressed, I felt increasingly like my muscles were getting weak when they shouldn’t be. It was hard to know if I was correct, because I always had some excuse. For example, my thumb would feel very weak but it was because I was scrolling on my phone too much. My arm was weak but it was because I was lying in the wrong position. Still, I always felt like the weakness was very disproportionate to my excuse for it.
As I drove up to visit my mother for Pascha, I was feeling like holding the steering wheel was making my arms and hands weak, and it was worrying me. I had beef sticks in what I had packed, and I had this intuitive sense they could make all this go away, but I really wanted to hold out until after church that night, and I did. As I sat down in church with my mom, she asked how I was, and I told her I was anxious because I felt like I had some muscular problem.
During Lent, there was a point where the anxiety was telling me anything I would try to fix the problem would make it worse, but I eventually cracked through this by reminding myself that this is actually my expertise and I have fixed problems like this many times before. This ultimately led me to supplement with my full rotation of B vitamins, including biotin and lipoic acid, but I do not remember for how long I supplemented them. I did feel like this took the edge off the problem and helped it reverse course rather than get worse, but it did not eliminate the problem, at least immediately.
The fact that muscular contraction is driven by acetylcholine and liver and eggs are the best source of its precursor, choline, made me suspect I was choline deficient. The best supplement to boost acetylcholine is alpha-GPC, but I had been taking that regularly during my focused work days. I do not remember if I added more of it along with the B vitamin cocktail.
After Pascha, I added back liver, egg yolks, dairy products, and meat, and the problem very rapidly resolved.
Soon after was when my allergies started getting bad, which I fixed with a supplement cocktail and an emphasis on making sure I was hitting minimum targets for egg yolks and liver, as described in What I’m Doing for Allergies. As the pollen I was allergic to subsided, and the allergies seemed 100% gone, I eliminated most of the supplements, but I continued to eat three eggs every day and supplement with the equivalent of once ounce of liver every day.
I have since avoided any type of fasting until I find a precise solution to this problem and enable my body to tolerate it, which I believe I am on the brink of now.
An interesting aside that ties back to my use of coconut oil to help my fasting intolerance during grad school: initially during Lent I was trying to avoid added oils, but partway through I started adding coconut oil to all my meals. This caused a spontaneous weight loss of five pounds. Perhaps I just needed more fat, but my sense was that the medium-chain fatty acids in the coconut oil were benefitting my metabolism.
Peripheral Neuropathy While Under-Eating
The icing on the cake occurred on August 10.
The month leading up to this day was full of extreme overwork. I felt that the one thing I needed to do to completely clear COVID off my plate so I could focus exclusively on my Vitamins and Minerals 101 book was make a final update to my COVID Guide (free to Masterpass members here) that included a protocol for long-COVID, and publish a Healing From COVID Vaccine Side Effect guide (free to Masterpass members here).
I wound up working through four weekends in a row and taking them all off back-to-back once the guides were finished for a road trip (a model I’ve found to lead to much greater productivity and more effective rest, and have largely continued since then).
In the days of the final stretch, however, I was not simply working through weekends but was often on the computer for 15 hours or more a day and forgetting to eat.
I got invited to a get-together with anti-mandate friends at a brewery the day I released the vaccine guide, August 10, at 7 PM.
I finally ate breakfast at 4 PM, and it was a salad with three medium-boiled eggs and a drizzle of olive oil, overall quite low in calories.
I thought everyone was going to be eating at the event, so I didn’t want to eat more until I got there.
As I was driving, I started feeling a collection of sensations I was wrongly at the time associating with “hypoglycemia,” and which were driven instead by under-eating. One was increased anxiety, and another was an empty feeling in my lungs, with a declining rate and volume of breath, which I believe is driven by low levels of carbon dioxide as my metabolic rate slows. If CO2 levels drop, the brain will make sure they are kept up by breathing less so that less CO2 is lost.
Part of me felt like I should eat some snack bars I had in the car, but the other part of me really wanted to save my appetite for a giant feast at the brewery.
Once I arrived, however, I realized that no one was eating! Everyone was just drinking a beer!
I knew I would need to eat sooner rather than later but, when in Rome…
So I got a beer and got into very lively conversation. The more and more excited I became and the more and more I talked, the more I felt the emptiness in my lungs increase.
Then my hands and feet started tingling. Badly. As if I was lying on them for a while only there was no such physical pressure to explain it.
At that point I ordered some food.
Once it arrived, I scarfed it down quickly, and my hands stopped tingling almost instantaneously.
However, my feet continued to tingle. I had a feeling it was from sitting on a hard bench, but it felt awkward to stand when everyone I was talking to was sitting. Eventually I did stand up and this made the tingling in my feet slowly resolve.
In the months after this, I found the tingling would sometimes come back, usually when sitting for a while, but never anywhere near as strongly. I would often wake up with my hands feeling a little numb, but this came and went in waves.
As much as I can easily attribute the major tingling event to the elevated stress of finishing a project, the overworking, and especially the under-eating, there was some persistence of the problem.
Regardless, this was the second neurological concern of the year, and my sense was that this problem was connected to all of the other vulnerabilities I have described above, including the negative reactions to veganism and terbinafine. It was time to finally get to the bottom of it.
Running the Labwork
If my current self were to talk to my 2018 self, I would tell my 2018 self to run the comprehensive nutritional screening that I developed for Testing Nutritional Status: The Ultimate Cheat Sheet (free to Masterpass members here).
Instead, I largely followed the “cost-saving” approach, focusing on dietary analysis and only running specific labwork when there were reasons justifying it.
I believe this was a big mistake and wasted four years of not getting to the bottom of my main biochemical weaknesses.
Nevertheless, in August of 2022 I was still in cost-saving mode, as I was worried that spending too much money on anything would deplete my savings and make me have to focus on making money instead of finishing my book. So, I did an “insurance-friendly” version of the comprehensive screening that left out plasma amino acids and urinary organic acids but included nutrient concentrations in blood and blood cells from LabCorp. These results are in the footnote.1 For the rest of this article, other lab results are also included in the footnotes associated with their description in the main text.
Biotin As the Major Problem
The most out-of-range nutritional marker was that my serum magnesium was high. Yet the only supplement I take with magnesium is Just Thrive vitamin K2, which contains a measly 36 milligrams. The doctor who ordered the test forgot to order red blood cell magnesium. These two markers should never be ordered one without the other, as the only way they can be rationally interpreted is when ordered together. The most likely explanation for the high magnesium was that my blood cells were not taking it up effectively.
The lowest marker was my uric acid, which I threw in as a possible proxy for molybdenum status, since molybdenum is needed to make uric acid. It was right at the bottom of the range. I am currently awaiting other results that will help me confirm if using the marker in this way worked, and I will write about this in the future. Legumes and liver are the best sources of molybdenum, and while I was getting supplemental liver I was not eating legumes, so if my molybdenum was running low it wouldn’t be very surprising.
On the other hand, the marker most wildly discrepant with my diet was the biotin (vitamin B7). My biotin was in the bottom 10.25% of the range, even though the most reliable sources of this vitamin are liver and egg yolks, and my intake of these has to be in the upper 95th percentile of the population.
Moreover, if my levels were this low after systematically getting three egg yolks and one ounce worth of supplemental liver per day for months, they must have been below the normal range during Lent when I was not consuming these.
This enormous discrepancy between my blood and dietary levels of biotin caught my attention as the candidate that could most parsimoniously explain most of the other results, could explain what must have been worse during Lent, and could lead me to this holy grail of a simple genetic explanation for my intolerance of fasting and animal food deprivation.
Since biotin is needed for production of ATP, the major cellular energy currency, it could explain why the serum magnesium was high (magnesium is trapped inside cells by chelation with ATP), why my uric acid was low (turning molybdenum into its enzymatic cofactor form requires ATP), why my copper was low-normal (its transport requires ATP), and why my B6 was the B vitamin that was next closest to the bottom of the range (its retention in the body is driven by ATP-trapping).
As described in When High-Dose Biotin is Truly Needed, biotin deficiency causes immune dysfunction that includes in some people deficient immunoglobulins, and this perhaps could explain why my immunoglobulins were low-normal, driving the albumin-to-globulin ratio to be flagged as high. As described in High Protein? You Need More Biotin, the glucose running slightly high might have been caused by biotin deficiency compromising the activity pyruvate carboxylase, an enzyme needed for glucose metabolism.
Furthermore, any impairment in ATP production could easily explain why I am so intolerant of fasting and under-eating.
I Forgot All About My Chronic Conjunctivitis!
As I thought about biotin, I instantaneously connected it to my multiple experiences of fungally driven whole-body eczema, since this is one of the classical symptoms of biotin deficiency, but then I also realized that I had spent the last 20 years completely forgetting about my periods of chronic conjunctivitis!
Conjunctivitis is another classical symptom of biotin deficiency.
When I was a little kid I had conjunctivitis so often that I had internalized a protocol about what to do when I wake up with my eyes glued shut: stumble my way over to the bathroom sink, put warm water on my eyes for long enough to peel the lids apart, then make a hot compress with a washcloth and use it until the pink eye goes away.
While my memory from the period where I smoked marijuana as a teenager is poor, I vaguely remember having more episodes of conjunctivitis where I used eyebright tea internally and in a compress.
Another round of chronic conjunctivitis hit me during my first post-Weston Price episode of whole-body eczema. During the year it took me to solve the skin problem, I got conjunctivitis so regularly I developed a new protocol for it based on the internal use of chicken coconut soup, which I was convinced at the time had powerful immune-boosting powers. Notably, it is high in glycine from the chicken stock and medium-chain fatty acids from the coconut, two topics we will return to below. As far as I can remember, the conjunctivitis stopped when the eczema went away.
I developed severe conjunctivitis one other time that was somewhat close in proximity to the whole-body fungal eczema attack of 2017 as well. To the best of my memory, this was likely a few months before the attack of the eczema. My eyes were so photophobic that it caused extreme pain to try to look at my phone screen to find the phone number of an ophthalmologist. I took a bunch of vitamin A and anything else immunosupportive I could find in my cabinet and it went away before I got my ophthalmologist appointment the next day. Once I went in, he told me there was no way my supposed conjunctivitis was an infection if it went away as fast as I claimed, and that my problem was blepharitis and I needed to wipe my eyelids with chemicals every day and take omega-3 fatty acids. His advice made my eyelids burn so I forgot about it and moved on.
My Baby Eczema
Besides the conjunctivitis beginning in childhood, my problem with eczema actually began when I was a baby. This was blamed on food allergies, and I was deemed allergic to all kinds of foods. My mom brought me to a chiropractor for it when I was two, at which time the “allergies” went away, though I may have outgrown them.
That two classical signs of biotin deficiency — eczema and conjunctivitis — began in infancy and childhood is consistent with a genetic interpretation.
My Mother’s Hair and My Grandfather’s Hearing
As I was investigating the biotin angle, I wrote three major articles on biotin:
After reading the first article, my mom informed me that she was taking a supplement called Dermal Repair that has 2.5 milligrams of biotin, which is a very large dose that no one should need except as a short-term loading dose, unless they are the one in 30 people that I estimate have a genetic basis for needing high-dose biotin. She had been taking it for about two years and it had been the one and only thing that had reduced her life-long elevated rate of hair shedding to normal.
When she stopped it for four days to run lab tests, her rate of hair shedding increased. When she resumed it, the hair shedding went back to normal.
While there are other ingredients in the supplement, this is consistent with my mother having a genetic basis for a high biotin requirement.
My grandfather had hearing aids for my entire memory, probably starting in his 60s, though family say he may have needed the hearing aids much earlier than that and had been in denial of it. Since hearing loss is common in biotinidase deficiency, a genetic deficiency in the ability to recycle biotin, this could be consistent with my grandfather passing on such a deficiency to me through my mother.
This family history strikes me as supportive of a genetic basis for a high biotin requirement.
My Biotinidase Activity Was Low-Normal
A few weeks later, I ran some additional labs that were missing from my first batch and included a measurement of my biotinidase activity.2
My biotinidase activity was 23.6% into the normal range. While no one would diagnose me with a deficiency from this, it could be a contributor to why my biotin levels were so discrepantly low compared to my diet.
These labs also confirmed that my red blood cell magnesium was normal, not high like my serum suggested. My red blood cell magnesium was not low, however, so this suggests some very mild impairment in cellular retention.
Generationally Compounded Biotin Deficiency
One interesting thing to note here is that a deficiency in biotin recycling should theoretically lead to generationally compounded biotin deficiency down maternal lines, with males interrupting the compounding and offering respite to the next generation.
This is true even though the inheritance is autosomal recessive, which predicts no sex differences and an inheritance pattern typical of blue eyes.
However, the use of the word “recessive” here is an extremely misleading artifact of the reality distortion imposed on the issue by the medical diagnostic model. In reality, carriers have half the biotinidase activity as non-carriers. This makes them “normal” in the reality distortion filter of medical diagnosis. However, when looked at using a systems biology approach, where we focus on the interactions between continuous variables and the resulting epiphenomena, it makes them run a smaller biotin deficit for decades, which should ultimately make them wind up with symptoms much later in life that no one will ever diagnose.
The reason I predict the generational pattern is that the mother provides the baby with a biotin pool at birth, and the deficiency symptoms will not set in until the deficit between dietary supply and metabolic demand leads to the depletion of that pool. Even in a severe deficiency this can take years. In a mild deficiency, it could take decades.
If my grandfather had deficient biotin recycling, he could have passed that gene to my mother. If she had deficient biotin recycling, she would become progressively more biotin deficient leading up to becoming pregnant with me. This would leave me with a smaller biotin pool at birth than someone born to a mother without such a deficiency. I would thus have worse symptomology with earlier onset than my mother or grandfather even if my father made no contribution to my biotin genetics.
On the other hand, even if I never figured this out, if I had children with a woman who did not have such a deficiency, I would interrupt the inheritance of the depleted biotin pool, since my personal biotin status would have nothing to do with the biotin pool left to my children at birth. I would contribute the gene for poor recycling, but not my depleted biotin pool.
Self-Experiments and Other Lab Work
While I had a strong hunch about the biotin, I did not yet have any functional markers showing biochemical effects clearly caused by biotin deficiency.
There were two points that gave my hypothesis a problem:
The one time I had ever run urinary organic acids was as part of a Genova ION panel ordered by an environmental medical doctor in 2017.3 The one biotin deficiency marker, beta-hydroxyisovalerate, was not only normal, but in the second quintile, which is the lower half. Were I biotin deficient at that time, it should have been high.
If biotin was what cured my veganism-induced mental problems, why did I develop classical biotin deficiency signs — conjunctivitis and whole-body eczema — after that recovery?
Thus, if this really was the holy grail of a simple genetic explanation of my lifelong health history that could lead to the personal greatest biohack of all time, I needed better evidence.
This evidence took two forms:
Self-experiments testing the metabolic effects of fasting, and testing whether I had signs of a pyruvate carboxylase deficiency secondary to biotin deficiency, especially whether I had paradoxical rises in ketones after eating carbs, and whether I had glucose intolerance that could be mitigated by co-ingestion of protein. The rationale for this is described in High Protein? You Need More Biotin. I will cover the results of these experiments in the next installment.
A search for functional markers indicating real biochemical effects of biotin deficiency occurring in my body. I will describe these below.
At this point my concern about money went out the window. Ensuring my health was my top priority.
Moreover, I soon realized that if I’m running this many tests, I might as well run all the tests so that I could use my experience to help you, my readers, better navigate what nutritional tests are worthwhile.
ION, NutrEval, OAT, Acylcarnitines, and More
If you do not have a background in biochemistry, much of this might be difficult to understand, in which case I recommend skipping this section in the short-term and taking my Energy Metabolism class in the medium-term. If you choose to skip this section, skip ahead to “Putting It All Together.”
The first step was to repeat the Genova ION panel4 to look for biotin-specific markers and to run a Genova Methylation Panel5 to look for the low-ATP pattern I described in How Energy Deficiency Hurts Methylation.
The methylation panel showed that my homocysteine was proportional to my methionine, but that my SAM was flagged as low, my SAH was high-normal, and my SAM was low relative to methionine while my SAH was high relative to my homocysteine. This confirmed the low-ATP pattern.
The ION results were far more complex.
On the one hand, the beta-hydroxyisovalerate marker of biotin deficiency was still not elevated. In fact, it was in the first quintile, even lower than in 2017. Nevertheless, 10% of people made experimentally biotin deficient never develop an elevation of this marker, as I covered in High Protein? You Need More Biotin.
On the other hand, my aspartate was flagged as low. Aspartate is primarily synthesized from oxaloacetate using the B6-dependent “liver enzyme” aspartate aminotransferase (AST), and the oxaloacetate is made primarily from pyruvate (derived in turn from glucose), using the biotin-dependent enzyme pyruvate carboxylase. In moderate genetic defects of pyruvate carboxylase, aspartate is usually low or undetectable.6 The facts that my B6-specific markers (xanthurenate, kynurenate, and quinolinate) were not elevated and that there was no indication of a broad-based transamination problem supports that this was due to poor pyruvate carboxylase activity, plausibly attributed to biotin deficiency.
Further, my odd-chain fatty acids were all at or near the top of the range. These are indicators of biotin deficiency because biotin is needed for their downstream metabolism as a cofactor for propionyl CoA carboxylase.
The low aspartate conflicts with my 2017 panel, which showed aspartate high-normal. However, the overwhelming abnormalities on that panel were the low glutamine, high glutamate, and high glutamate-to-glutamine ratio, which indicate a problem with acidity. This was the major way I identified my need for bicarbonate. Acidity causes the hydrolysis of glutamine to form glutamate, thus liberating ammonia that can be used to buffer the acid. I suspect the aspartate was high normal not because my pyruvate carboxylase was in good shape, but because the acid burden was leading to an analogous hydrolysis of asparagine as an alternative way of forming aspartate.
My 2022 panel shows the glutamate-to-glutamine ratio is bottomed out, indicating complete solution of the acidity problem, thus unmasking the low aspartate.
Three of my odd-chain fatty acids were similarly high normal in 2017, indicating a persistent problem in that pathway.
The high-normal alpha-ANB, which is driving the alpha-ANB-to-leucine ratio to be flagged as high, reflects a backup of valine and methionine metabolism that similarly feeds into propionyl CoA carboxylase just like the odd-chain fatty acids. The alpha-ketoisovalerate is flagged as high and this similarly could reflect a backup of valine metabolism due to deficient propionyl CoA carboxylase.
The alpha-ANB wasn’t measured on the “New York” version of my ION in 2017, and none of my branched-chain amino acid metabolites were elevated, but I do not remember how much protein I was eating at the time of the 2017 ION so the discrepancy might easily be resolved with differences in protein intake.
A deficiency of pyruvate carboxylase should compromise activity of the citric acid cycle, leading to an elevated ratio of NAD+ to NADH, with some of that being rescued by using NADPH using the enzyme NADPH transhydrogenase, thus leading to a decline in NADPH levels. This could hurt the recycling of BH2 to BH4 and thus lead to the high ratio of phenylalanine to tyrosine.
The methylation panel showed my glutathione was borderline low, while the ION panel showed my alpha-hydroxybutyrate was elevated and my isocitrate is low. This probably reflects low ATP levels impairing glutathione synthesis, leading to increased catabolism of homocysteine to form cysteine used for glutathione sysnthesis, and leading to mild oxidative stress inhibiting aconitase, which forms isocitrate and is the enzyme of the citric acid cycle most sensitive to oxidative stress. Nevertheless, this mild oxidative stress is not leading to oxidative damage, as shown by the mid-normal levels of lipid peroxides and 8-hydroxy-2-deoxyguanosine, a marker of oxidative damage to DNA.
The elevated ratio of hydroxyproline to proline is probably driven by bone broth consumption and thus uninteresting.
While it is interesting that my vitamins A and E levels were elevated here, this seems to be a fluke, as neither was elevated in my recent LabCorp bloodwork, nor in the Vibrant America panel I got soon afterward.
The next step was to run a Great Plains Organic Acids Test to see if my methylcitrate was elevated.7
It was not. In fact, it was on the low side of normal.
Methylcitrate is formed as a detoxification product of propionate using oxaloacetate. Propionyl CoA acts as an alternative substrate to acetyl CoA for use in the citrate synthase reaction at the beginning of the citric acid cycle. However, if biotin deficiency compromises pyruvate carboxylase activity, the oxaloacetate will not be available to form methylcitrate. This thus seems like a very poor marker of biotin deficiency to me and its absence has an easy explanation.
Consistent with the ION panel, my alpha-hydroxybutyrate was elevated.
In addition to the ION, it showed my 3-hydroxyglutaric acid was elevated. I will return to this in the genetics section.
In contrast to the ION, my peroxisomal markers (suberate and sebacate) were mildly elevated and my urinary ketones were wildly through the roof. My beta-hydroxybutyrate was 13 times the top of their range and my acetoacetate was nearly 16 times the top of their range. My acetoacetate-to-beta-hydroxybutyrate ratio — which they do not calculate — was 6.3. Normal ratios (based on a small amount of data since normal ranges for the ratio are difficult to find) appear to be between 1 and 3. The dominance of acetoacetate over beta-hydroxybutyrate is consistent with an elevated ratio of NAD+ to NADH resulting from poor pyruvate carboxylase activity, and ketones being through the roof is consistent with a lack of oxaloacetate from poor pyruvate carboxylase activity.
But why didn’t the ION show this?
I have an explanation, but I hadn’t discovered it yet. So we will return to this.
Since my biotinidase activity was low-normal but not flagged low, I was still in search of other explanations for my discrepantly low biotin, and the one other explanation would be a defect in biotin transport. There are no commercially available tests for intracellular biotin or for dynamic biotin transport. However, one of the transporters for biotin is the sodium-dependent multivitamin transporter (SMVT), which also transports pantothenic acid and lipoic acid. The Vibrant America Micronutrient Panel has extracellular and intracellular pantothenic acid, so I ran that to look for evidence of an impaired SMVT. I did not find any such evidence. I described this, as well as a Spectracell I ran simultaneously, in Vibrant Vs. Spectracell Panels and the data are included in the footnotes on that post.
My last round of labwork before starting biotin supplementation was to see if I could detect this putative propionic acid backup — masked when looking at methylcitrate by my putatively low oxaloacetate — using 3-hydroxypropionic acid on the NutrEval8 and using propionylcarnitine on an acylcarnitine profile.9
The 3-hydroxypropionic acid on the NutrEval was low-normal, arguing against my hypothesis.
However, the propionylcarnitine was, while in the normal range on the acylcarnitine profile, present, which should in an of itself be seen as significant.
The acylcarnitine profile represents detoxification products of metabolic intermediates where carnitine brings those intermediates out of the cell, into the blood, for excretion into the urine precisely because their metabolism inside the cell is impaired. The reason the normal range for propionylcarnitine includes zero is because it is normal, and, almost certainly optimal, for the concentration of this substance to be zero.
Thus, the acylcarnitine profile does not really “confirm” my hypothesis but it is very consistent with the high-normal odd-chain fatty acids and alpha-ANB, and the flagged high valine metabolite from the ION. The NutrEval confirmed the elevations of odd-chain fatty acids (even flagging several in the red), flagged my valine as yellow, and showed most branched-chain amino acid metabolites as high-normal, this time flagging only the leucine metabolite isovalerylglycine as yellow. Thus, there is broad agreement across tests of a moderate backup in propionyl CoA carboxylase.
The NutrEval showed glutaric acid acid as slightly in the yellow range. This and the 3-hydroxyglutaric acid from the Great Plains test are lysine metabolites, and my NutrEval flagged lysine as high.
The NutrEval did not confirm the markers of mild oxidative stress on the ION, suggesting this is not a consistent or dominant problem for me.
The NutrEval showed my aspartate as undetectable, confirming the low aspartate on the ION.
Neither the ION nor the NutrEval measure urinary acetoacetate, so I could not calculate the ratio of ketone bodies with either of these like I could using Great Plains, but my beta-hydroxybutyrate was flagged as high. However, it was only 3.7 millimoles per liter, nearly seven times lower than the Great Plains figure of 25, yet it was 3 times higher than the ION figure, which, after adjusting for the unit differences, would be 0.87.
Again, I will resolve this discrepancy in ketone measurements shortly.
The low or undetectable aspartate and the inappropriately elevated ketones indicate to me that my pyruvate carboxylase activity is impaired much more strongly than my propionyl CoA carboxylase activity, and that this explains why the Great Plains could not detect an elevation of methylcitrate.
However, the acylcarnitine profile suggested something else entirely had much more significance than the backup in propionyl CoA carboxylase. I will discuss this in the (V)LCAD section below.
First, let’s resolve these conflicting ketone reports.
My Biotin Status Had Been Declining All Year
From September through the present, I “froze” my diet and supplements to reflect what I had been eating through the summer so that I could study my metabolism in the putatively biotin-deficient state without confounding from an evolving input of nutrients.
In my mind, this meant my biotin status was going up all year, because it had dipped the most during Lent when I was not eating liver or egg yolks.
During this period of time, I developed two classical signs of biotin deficiency that I had not had in years. One was conjunctivitis. I cured it rapidly by putting contact lens solution in my eyes and it never came back. Another was an itchiness behind my right knee, in the exact spot my fungal flare from 2017 was worst. Topical antifungals got rid of it.
I was confused, because while this lined up so perfectly and poetically with the hypothesis I was chasing, it made no sense that my biotin deficiency would be getting worse.
Lo and behold, that is exactly what was happening. The day before I began biotin supplementation, I measured my serum biotin again.10 It had fallen in half, and was now in the bottom 2.5% of the normal range rather than the bottom 10.25%.
This means that my biotin status was in decline all year long. Suddenly, the conflicting ketone measurements on the ION, Great Plains, and NutrEval made sense.
The ION was taken September 9, and the Great Plains was taken December 20. During the course of that time, my biotin status declined and my pyruvate carboxylase activity deteriorated, shooting my urinary ketones through the roof and leading to an extremely oxidized ratio of ketones.
The NutrEval was taken on February 4, when my biotin status had continued to deteriorate, as shown by the aspartate becoming undetectable instead of just flagged low. However, the reason the ketones moderated is that I had been doing intensive glucose tolerance tests on myself in the month leading up to the NutrEval.
Thus, my pyruvate carboxylase activity had been deteriorating the whole time, but my ketones were only wildly through the roof in the absence of blasting myself with dextrose powder, and in the presence of blasting myself with dextrose powder my ketones were merely out-of-range high.
I will have to confirm these three tests have comparable urine ketone measurements by using the same urine simultaneously in my future experiments, but for now my tentative conclusion is that the apparent conflicts can be reconciled by taking into account my deteriorating biotin status and my glucose tolerance tests.
The results of the glucose tolerance tests and other self-experiments will be described in the next installment.
The declining biotin status leads to the dramatic conclusion that my diet has not at all been what is keeping my biotin in the normal range, but rather my past use of biotin supplements is.
Presumably, there is one great arc of declining biotin status since birth, as my impaired biotin recycling leads to a progressive depletion of what I was born with.
Then, there are smaller arcs that last years that are driven by months of biotin supplementation at a time. My iHerb orders show that I bought a bottle of Jarrow biotin, 5 miligrams, 100 capsules, on November 4, 2019, and then purchased Life Extension biotin, 600 micrograms, on December 13, 2020. I do not remember if I purchased any other biotin at a local store during that time. So at minimum I loaded biotin at 5 milligrams per day for 100 days, and then occasionally used 600 micrograms per day intermittently over the course of the following two years, until I did the last round of it after Lent in 2022.
I suspect that it was this biotin that accounts for why I was still just barely in the normal range at the beginning of last month.
A Fatty Acid Oxidation Problem
One thing the acylcarnitine profile made abundantly clear is that I had an entirely different problem with fatty acid oxidation that had not occurred to me at all until I ran the test.
Whenever you run such a profile, you need to ignore the useless and harmfully misleading statement at the top, “THIS PATTERN OF MILDLY ELEVATED ACYLCARNITINES IS NOT CONSISTENT WITH A SPECIFIC DISORDER.”
As stated in the 2022 edition of the Saudubray textbook, “. . . in contrast to the usual belief, a normal or nonspecific urinary [organic acid] and acylcarnitine pattern, even at the time of an acute attack, does not exclude an inherited FAO disorder.”
The appropriate use of such a test is to look for the patterns that emerge, and, if needed, to use various dietary provocations to analyze the changes in patterns like a biochemical detective.
The logic of an acylcarnitine profile is as follows.
An acyl group is what you get when you join an acid to another molecule. In biochemistry, acids are usually joined to carriers in an ester bond. Coenzyme A, or CoA, is a carrier derived from pantothenic acid (vitamin B5), that carries fatty acids as well as some amino acid derivatives through metabolic pathways while they are being burned for energy. Carnitine is needed to transport some of these acids, such as long-chain fatty acids, into the mitochondria so this can take place. However, carnitine can also be involved in exporting them and their byproducts.
When a CoA-requiring metabolic pathway is impaired by a genetic defect, nutrient deficiency, or toxin, the intermediates will hog the CoA pool. Essentially, the passengers refuse to get off the CoA bus, and there are no empty buses to pick up new passengers. This CoA starvation will, if it is not resolved, cause an abrupt halt to all metabolism. Since this would result in death, the block must be resolved.
Further, small acids such as propionic acid or methylmalonic acid are similar enough in size that their CoA esters — propionyl CoA and methylmalonyl CoA — will resemble acetyl CoA and act as an imposter of acetyl CoA, thereby inhibiting processes that depend on it. One well characterized effect is the inhibition of N-acetyl-glutamate synthesis, which is the on-switch for the urea cycle. Thus, disorders that lead to CoA starvation, and especially those that lead to the accumulation of alternative small esters of CoA, lead to secondary hyperammonemia.
In order to resolve this, carnitine and glycine are used to free the CoA by acting as alternative carriers. Thus, acyl-CoAs become free CoA and acylcarnitines or acylglycines. These ultimately are needed to get the acyl groups out of the cell and into the blood, and from there into the urine. Acylcarnitine profiles or acylglycine profiles help you see which specific acyl groups have been detoxified in this manner.
Organic acids from these pathways rarely leave cells by themselves. Their transport is often dependent on carnitine. Cell experiments show that a carnitine deficiency will mask the organic acid markers of biotin deficiency by preventing the organic acids from leaving the cell. This suggests that the organic acid test would fail to show the markers yet the cellular toxicity would be greater.
For all these reasons, acylcarnitine and acylglycine profiles are used in the diagnosis of many inborn errors of metabolism, and high-dose L-carnitine and L-glycine is used in the treatment of most inborn errors in CoA-requiring pathways. The reason L-carnitine is used instead of acetyl-L-carnitine is because it has its acyl-carrying site open and ready to bind metabolic intermediates rather than being already bound by an acetyl group.
However, there is some controversy over whether L-carnitine is always beneficial, because there is some evidence that acylcarnitines themselves can contribute to heart arrhythmia. Therefore, such treatments need to be vetted for their benefits and measurements should ensure they are leading to the clearance of metabolic intermediates into the urine and not the accumulation of their esters in the blood.
Of all the acylcarnitines, only acetylcarnitine has a lower bound of the normal range substantially above zero in plasma, and low-normal concentrations of it might reflect the need for carnitine transport rather than detoxification of acetyl groups. Virtually every other acylcarnitine has a normal range that includes zero.
My acylcarnitine profile showed a substantial presence of branched-chain amino acid intermediates (isovaleryl/2-methylbutyryl, tiglyl/methylcrotonyl, hydroxy-isovaleryl), as well as the propionylcarnitine I was looking for, and an accumulation of acetyl and malonyl that I suspect may reflect a lack of NADPH needed for fatty acid synthesis, secondary to pyruvate carboxylase lowering NADH and NADPH transhydrogenase helping to compensate.
It also showed that glutarylcarnitine was at the exact top of the normal range, consistent with the glutaric acid-related markers on the Great Plains and NutrEval.
However, the only two markers actually flagged high are 14-carbon fatty acid-related markers, C14:0 (tetradecanoyl) and C14:1-OH (OH-tetradecanoyl). These are derived from fatty acid oxidation.
There are no direct markers of pyruvate carboxylase activity on an acylcarnitine profile, so this does not necessarily suggest the fatty acid oxidation problem is greater than the pyruvate carboxylase problem, but it does imply that it is greater than the problems I have with branched-chain amino acids and odd-chain fatty acids from the backups in the biotin-dependent enzymes specific to those pathways.
If I remove the markers related to branched-chain amino acids, odd-chain fatty acids, and fatty acid synthesis, and include only the ones relevant to fatty acid oxidation, this is what my profile looks like as a percent of the normal range:

Fatty acids are progressively oxidized from long to short by chopping off two carbons at a time. This chart suggests the biggest block is at C14. The catch is that any fatty acid longer than that will have to eventually hit the C14 point.
Looking at this chart alone, I would conclude that the fatty acids I can best tolerate are those shorter than ten carbons. That would include butyrate (C4) as well as caprylate (C8). The fact that C10 acylcarnitine is 3.5 times further into the normal range than C8 suggests that I would do much better with C8-specific MCT oil rather than the standard C8/C10 combo or than with coconut oil.
The natural food highest in butyrate is butter, but butter is mostly long-chain fats.
The natural food highest in C8 is coconut oil, but coconut oil is also mostly long-chain fats (the C12 fat lauric acid is often misclassified as medium-chain but it behaves like a long-chain fat in many respects) and has only 15% of its fatty acids as a combination of C8 and C10.
The oil I would tolerate best according to this chart is C8-specific MCT oil. And thus it is remarkable to me that my experience of spontaneously losing five pounds when adding coconut oil during Lent motivated me to buy Bulletproof Brain Octane C8 MCT Oil on June 25, 2022. A friend of mine visited nearly immediately after this who was accustomed to making Bulletproof Coffee in a Vitamix, so we made C8-coffee (no butter) together for a few days and drank it in the outdoor sunshine over some great conversations.
I didn’t try it long enough to see how it impacted my health, because I prefer cream in my coffee and quickly went back to that habit. I tried the C8 oil on salad, but it gave me loose stools. I believe I need to blend it into a drink to be able to absorb it better, a test that will remain for the future.
The question arises which fatty acid oxidation enzyme is defective. There is a sizeable family of riboflavin-dependent acyl CoA dehydrogenases, some involved in amino acid oxidation and some involved in fatty acid oxidation. The main ones involved in fatty acid oxidation are short-chain, medium-chain, long-chain, and very long-chain acyl CoA dehydrogenases (SCAD, MCAD, LCAD, and VLCAD), as well as acyl CoA dehydrogenase 9 (ACAD9). ACAD9 plays a dual role in oxidizing fatty acids and helping to assemble complex 1 of the mitochondrial respiratory chain. Thus, its deficiency can cause both a fatty acid oxidation disorder and a respiratory chain disorder (see here and here).
ACAD9 seems to be involved in modifying fatty acids, as it often skips over certain chain lengths. For example, it will chop two carbons off the omega-3 fatty acid DHA to form EPA, but it will not further metabolize EPA. It will break oleic acid (C18:1) down to C14:1, but it will skip over the breakdown of C14 and C12 altogether. But then it will break down fatty acids with 9, 10, or 11 carbons. This seems to imply it plays an important role in remodeling fatty acids for the synthesis of various structural compounds.
ACAD9 is responsible for making the C14:1 that appears in VLCAD deficiency and its deficiency causes virtually no C14:0 acylcarnitine. This is wildly at conflict with my profile, where C14:0 is the peak, and where C14:1 is present, and where C14:1-OH is the second highest acylcarnitine.
While this figure is from rat liver, I cannot find such an elegant comparison of the human enzymes, so I will include it here:

From among SCAD, MCAD, and LCAD, my acylcarnitine profile clearly most closely conforms to LCAD.
However, LCAD deficiency has strongly overlapping specificity with VLCAD. For example, when human VLCAD-deficient cells are incubated with oleic acid, their greatest disruption is at the breakdown of C14:

The tissue distributions of VLCAD and LCAD are different. They are both found abundantly in liver, heart, and muscle, but LCAD is more important in kidney, prostate, lung, and thyroid, whereas VLCAD is more important in adrenal gland, small intestine, and uterus.
The 2022 edition of the Saudubray textbook lists LCAD in “other potential defects” saying that its deficiency has been reported in two cases of sudden infant death but that “the paucity of cases may be due to the overlapping substrate specificities of LCAD and VLCAD.”
I suspect instead that this is ascertainment bias created by the dominance of VLCAD in skin fibroblasts and the widespread use of skin fibroblasts to assess enzymatic activity in human patients.
Thus, I believe my second problem is either a LCAD or a VLCAD deficiency, that this is a greater problem than my biotin-related propionyl CoA carboxylase deficiency, but perhaps not as great a problem as my biotin-related pyruvate carboxylase deficiency.
The Carnitine Problem
The presence of abnormal acylcarnitines raised the question of whether the need to detoxify metabolic intermediates could be depleting me of carnitine, and thus masking some of the biotin-related organic acids.
I got the acylcarnitine profile back soon after I started supplementing biotin, and then realized it did not have a measure of my total and free carnitine. So, I ran total and free carnitine at LabCorp11 after I had spent a few days taking 600 micrograms or 1.2 milligrams of biotin per day.
These results show that my total carnitine is 8.6% into the bottom of a very broad normal range, while my free carnitine is 16.7% into the bottom of its range.
If the carnitine were over-occupied by the detoxification function, we would expect the esterified fraction to be high, but in fact it is only 22% into the bottom of the normal range.
However, I did not measure any of this in urine, and I think it is very possible that the selective urination of acylcarnitines is what is bringing down my free and thus my total carnitine in plasma.
The converse would be to believe that I have a primary deficiency of carnitine. However, this seems wildly at odds with my urinary ketones blowing through the roof when biotin deficiency is present and glucose loading is not.
Thus, my current tentative belief is that the low-normal carnitine levels are a consequence of using carnitine to detoxify metabolic intermediates, and that this is compromising the detection of some of the biotin-related organic acids.
Genetic Investigations
My first approach was to search my 23andMe data using Promethease.
This led me to identify heterozygous status for rs34885143(A;G) in biotinidase, the enzyme that recycles biotin. This is classified as pathogenic, with arguments that it should be reclassified as benign. Its frequency is less than 0.4%. This may explain my low-normal biotinidase activity.
This also led me to identify homozygous status for rs8012(G;G) in the enzyme responsible for glutaric aciduria, a defect in lysine which has been found to increase glutarylcarnitine. This is likely solely responsible the mild elevations in glutaric acid and lysine-related markers across the Great Plains test and the NutrEval.
One fascinating connection I find to this is that the Saudubray textbook lists temporal lobe hypoplasia (underdevelopment) as one of the primary consequences of severe glutaric aciduria. I have long suspected that my difficulty using faces to identify people (I usually rely on abstractions such as what we talked about to identify people I don’t know well) could represent some underutilization of my temporal lobe. Perhaps this very mild deficiency in glutaric acid metabolism is responsible for very mild temporal lobe hypofunction in me.
After searching Promethease, I ran Dante’s Whole Genome Sequencing, and this additionally identified heterozygous status for the rs115532916 SNP in ACAD9 in their metabolic panel. Although this is rare (frequency less than 0.8%) and has “conflicting interpretations of pathogenicity,” (meaning it might be pathogenic), for the reasons I described above I cannot reconcile my acylcarnitine profile to an ACAD9 deficiency.
Masterpass member Sergey Andryukhin allowed me to run my Dante data through a private app he created but has not made available to the public called Gene Inspector.
This allowed me to identify a rare intronic enhancer in propionyl CoA carboxylase of unknown clinical significance:

This offers additional clarity to why my pyruvate carboxylase activity looks so much worse than my propionyl CoA carboxylase activity, despite having thus far found no genetic alterations to the former. If the latter can be expressed more easily and to a greater degree, then it could “steal” biotin from pyruvate carboxylase under conditions where biotin is scarce and my protein or odd-chain fatty acid intake puts pressure on propionyl CoA carboxylase to get moving.
Unfortunately, even with Gene Inspector all I can say for LCAD and VLCAD are that there are some rare SNPs with unknown consequence, but no direct evidence of a problem.
Nevertheless, I believe I at least have have carrier status for a genetic defect in one of these.
Why genetic?
Apart from “wanting” to find something simple with high explanatory power as described above, the fact that there is no indication of SCAD or MCAD deficiency argues for LCAD or VLCAD genetic deficiency. The reason is these are highly similar enzymes with similar cofactor requirements. Riboflavin deficiency causes defects in all of these enzymes, not just one of them.
Unfortunately I am realizing only now that my preferred original batch of labwork is missing whole blood riboflavin. Vibrant America uses serum and white blood cells, and those results do suggest I could use more riboflavin, but again were this problem driven by a deficiency per se I would expect a broad-based deficiency in fatty acid oxidation rather than a chain length-specific deficiency, not to mention a more consistent depression of my glutathione levels from a deficiency in the riboflavin-dependent enzyme glutathione reductase. While the glutaric acid metabolites could be seen as another manifestation of riboflavin deficiency, I already have clear evidence for a homozygous SNP to explain them genetically.
I will be doing further genetic testing to see if I can find additional genes in accordance with my hypothesis and will eventually write about which tests I have found most useful for myself and believe would be most useful for others.
I will also try to more clearly characterize my riboflavin status before acting on the assumption of genetics and to see if I simply need a little more riboflavin than my diet supplies or if I need high-dose riboflavin to fix a genetic fatty acid oxidation problem.
Unsolved: Peripheral Biotin Uptake?
One remaining question for me is whether I have a problem taking up biotin into my cells.
Biotin transport is not very well understood. It is widely agreed that the sodium-dependent multivitamin (SMVT) transporter can transport biotin, lipoic acid, and pantothenic acid. However, one case report identified a child who had a normal SMVT gene sequence and normal pantothenic acid transport, yet defective biotin transport in his blood cells. His parents had half the magnitude of defect as he had, strongly arguing for a genetic basis. This suggests there is a non-SMVT transporter that transports biotin. It may be that biotin is absorbed from the gut primarily using the SMVT but is transported into peripheral cells primarily using this other transporter.
Little followup has been done on this except to show that there is a non-SMVT biotin transporter in skin cells.
I tested my biotin absorption by weighing out 107 micrograms (1.5 micrograms per kilogram bodyweight) using a milligram scale and a Life Extension 600 microgram capsule, and then tested my absorption of it by testing my serum biotin before and 45 minutes after taking the supplement.
The main reason I did this is to see if I had a defect in intestinal absorption, or evidence of tissue depletion.
I modeled this after several studies done in the 1980s, here, here, and here.
Jess Thoene thought he had identified a patient with impaired intestinal absorption of biotin. However, Barry Wolf, who discovered biotinidase deficiency, asked him to retest the absorption after biotin loading. It was normal. Wolf’s explanation that the original finding of malabsorption was pseudo-malabsorption. The vitamin was never seen in circulation because the depleted tissues soaked it up instantly. Once the tissues were loaded with biotin, they no longer had such a voracious appetite, and the “absorption” went back to normal.
Using this principle, I followed the protocol of the third study, which had a larger sample size for the normal and defective plasma response.
Biotin peaks in serum at 45 to 60 minutes after taking it. Converting the LabCorp units to those reported in the above papers, my baseline serum biotin was 0.29 nmol/L, similar to the biotinidase deficiency patients, while my 45-minute serum response to biotin supplementation12 was a rise of about 4 nmol/L, similar to healthy adults.
This represents a paradox. Why is my baseline serum level similar to the patients with tissue depletion, yet my serum response to supplementation similar to healthy controls?
One possibility is simply that my tissues are not that depleted and that my serum levels are misleadingly low. However, another possibility is that I have the defective peripheral biotin transporter identified in the case report I discussed above, where tissue depletion is being masked by poor cellular uptake. Thus, the serum response appears normal not because the biotin doesn’t need to get into the cells but because it has great difficulty doing so.
If this is the case, it suggests that much of my biotin is lost in the urine no matter what I do, and that I need to maintain very high serum concentrations all the time to get my cellular concentrations to be normal.
If this is true, the evidence will be that biotin loading eventually makes my serum response supra-normal. That is, as the cells get less and less starving for biotin as my high-dose biotin progresses, the declining appetite unmasks their defective transport and my serum response becomes way higher than normal.
The only way to know is to test this by repeating the experiment after biotin loading.
Putting It All Together
My current model to understand my health over the last 41 years is as follows.
I have a genetically increased need for biotin that affects pyruvate carboxylase the most and propionyl CoA carboxylase the least. It is probably responsible for my recurring problems with eczema and conjunctivitis since childhood.
On my vegan diet, my biotin intake was much lower. However, it may be even more important that the products biotin is used to synthesize were also absent. For example, biotin is needed for the elongation of plant oil fatty acids into the essential forms of fatty acids found in animal products. Since lipoic acid is a modified fatty acid, it too is a byproduct of biotin-dependent fatty acid synthesis. Cholesterol and other lipids that make up myelin were also missing. Pyruvate carboxylase deficiency leads to demyelination, so the drop in biotin status and the lack of complex lipids from animal products acted as a double whammy to cause neurological catastrophe.
During my recovery, the desperate state of my nervous system took priority over the incoming biotin, lipoic acid, elongated essential fatty acids, cholesterol, and other products of biotin-dependent synthesis for a very rapid rebuilding and rejuvenation.
It may be that the nervous system always gets priority over the skin. Consistent with this, case reports of biotin-related genetic disorders often find that the skin rash takes the longest to clear. Or, it may be that cholesterol or another lipid in my diet acted as a catalyst to drive anything needed in concert with it into a symphony of neurological regeneration. Or, it may be that I have a defect in the non-SMVT biotin transporter that hurts biotin uptake into my skin more than it hurts biotin uptake into my nervous system. Regardless, my nervous system got first dibs on the biotin and my skin got it last.
My higher-protein, higher-fat diet put a greater tax on my fatty acid oxidation problem, my lysine oxidation problem (glutaric aciduria), and my need for biotin for the sake of metabolizing methionine, branched-chain amino acids, and odd-chain fatty acids. This led to substantial CoA sequestration and carnitine depletion during the attempts to detox metabolic intermediates to rescue the CoA pool. The taxing of the CoA pool would have worsened endogenous lipid synthesis, perhaps hitting the skin worst of all.
Thus, despite the rapid rejuvenation of my nervous system, the residual problems with biotin and the worsened problems with fatty acid oxidation led my skin to take over the experience of catastrophe.
While I still do not know whether the Primal Defense Ultra, the X-Factor Butter Oil, or both, cured the eczema, I would note that the butter oil likely concentrates butyrate and the Primal Defense may have helped my microbiome make butyrate, shifting the balance of fatty acids towards one that my cells could use. On the other hand, perhaps this shifted my microbiome somehow away from producing propionate, which I could not use. The butter oil may also have contributed meaningful amounts of arachidonic acid and other complex lipids beyond what I was getting from egg yolks and liver. And, of course, it could simply be that before my skin barrier could be completely fixed, inflammatory compounds from my gut were going systemic and inflaming my skin.
Due to the possibility that biotin may help detoxify oxalate, my biotin problem could explain my own oxalate intolerance, described in the oxalate article I just linked to. I would not rule out completely that I may have had some kind of oxalate dump from my brain that lit off my skin problem. However, this is not currently my lead hypothesis.
The fasting intolerance can be explained by inadequate energy production in the citric acid cycle from biotin not being available for pyruvate carboxylase and lipoic acid not being available for pyruvate dehydrogenase, alpha-ketoglutarate dehydrogenase, or branched chain alpha-ketoacid dehydrogenase, from biotin not being available for the production of ATP from branched-chain amino acids, methionine, and odd-chain fatty acids, and from long-chain fatty acids being poor sources of ATP. This could all cause hypoglycemia since ATP is needed for gluconeogenesis, or it could simply cause symptoms from lack of ATP itself.
The fact that I have multiple overlapping hits on mitochondrial ATP production makes me vulnerable to other assaults on my mitochondria, such as mold toxins, heavy metals, or anti-fungals, or to deficiencies of minerals such as calcium. ATP is needed to handle distribution of electrolytes and hydrogen ions across membranes. Without enough of it it, a low-calcium diet is more likely to cause tetany, barium toxicity is more likely to disturb potassium function, and acid-base balance is more likely to get distorted.
During Lent last year, I suspect that lower biotin and choline intakes both synergized with CoA sequestration to hurt my acetylcholine levels since biotin is needed to make citrate in the mitochondria to become the source of acetyl CoA in the cytosol, and CoA is of course needed to make acetyl CoA as well, and it is acetyl CoA that reacts with choline to make acetylcholine.
My repeated experiences suggesting a metabolic benefit of coconut oil probably result from it having the highest proportion of medium-chain fats, especially C8, in my diet. I am likely to benefit most from C8-specific MCT oil.
The glycine in the bone broth on my Weston Price diet or in that chicken coconut soup may have been helping to detoxify metabolic intermediates, and the totality of my animal products, especially all forms of meat, would supply more carnitine for the same purpose.
Moving Forward
I am currently in the middle of my first biotin-loading stage. I am keeping my diet and supplements otherwise constant so I can be certain what the biotin is doing.
I will test my “absorption” again to see if I have evidence that I have normalized my tissue status or have a defect in cellular transport that requires a much higher dose.
My default plan is to remeasure the functional panels after the biotin loading to see whether the biotin has normalized the markers of interest, especially the low aspartate, the high ketones, and the oxidized ketone ratio.
Providing this goes according to plan, step 2 will be to characterize my riboflavin status clearly and determine whether it makes sense to try a low dose before trying a high dose. My suspicion is I need a high dose to normalize the known defect in glutaryl CoA dehydrogenase, the enzyme responsible for the glutaric acid-related markers, and to normalize the putative defects in LCAD or VLCAD.
If the riboflavin is able to normalize the acylcarnitine profile completely, and if this leads to an improvement in my carnitine levels, it might all end there.
However, followup strategies in the event that does not happen may include replacing most of my dietary fat with C8-specific MCT oil to the point my digestive system can handle, or using high-dose carnitine or glycine to better detox metabolic intermediates, vetting these against the acylcarnitines and acylglycines in my blood and urine to make sure they are being effectively removed from my body.
The next installment in this series will cover the self-experiments I did in the biotin-deficient state, and then I will write about my experience biotin loading.
Did you have any cool insights while reading my story? If so, let me know in the comments!
Stay Immune Through the Winter
My new 7-page quick guide on how to not get sick this winter, Staying Immune Through the Winter, is free for everyone. All you need is a free or paid subscription to my Substack.
If the box above says “subscribed,” this means you are a Masterpass member and can download the guide here.
If the box says “upgrade to paid,” then you are a free subscriber and your guide is currently sitting in your inbox. Search your mail, spam, and trash for “staying immune through the winter” and make sure anything from Substack is going to primary.
If it allows you to enter your email address, do so, hit “subscribe,” and then find the email from “Chris Masterjohn, PhD” with the subject “Welcome to My Newsletter!” If you can’t find this, try searching for either of those terms and for “Substack.” At the top of the welcome email you will find your downloadable guide to not getting sick.
Join the Next Live Q&A
Have a question for me? Ask it at the next Q&A! Learn more here.
Join the Masterpass
Masterpass members get access to premium content (preview the premium posts here), all my ebook guides for free (see the collection of ebook guides here), monthly live Q&A sessions (see when the next session is here), all my courses for free (see the collection here), and exclusive access to massive discounts (see the specific discounts available by clicking here). Upgrade your subscription to include Masterpass membership with this button:
Learn more about the Masterpass here.
Please Show this Post Some Love
Please like and share this post on the Substack web site. This will help spread it far and wide, as subscriber likes drive the Substack algorithm. Likes from paid subscribers count the most, but likes from free subscribers come in the highest numbers and are the foundation for spreading the content.
Take a Look at the Store
At no extra cost to you, please consider buying products from one of my popular affiliates using these links: Paleovalley, Magic Spoon breakfast cereal, LMNT, Seeking Health, Ancestral Supplements, MASA chips. Find more affiliates here.
For $2.99, you can purchase The Vitamins and Minerals 101 Cliff Notes, a bullet point summary of all the most important things I’ve learned in over 15 years of studying nutrition science.
For $10, you can purchase The Food and Supplement Guide for the Coronavirus, my protocol for prevention and for what to do if you get sick.
For $10, you can purchase Healing From COVID Vaccine Side Effects for yourself or a loved one if dealing with this issue. It also contains an extensive well-referenced scientific review, so you can also use this just to learn more about my research into the COVID vaccines.
For $15, you can pre-order a single format of my Vitamins and Minerals 101 book, my complete guide to nutrition, which I am currently working full-time on finishing.
For $25, you can pre-order a digital bundle of my Vitamins and Minerals 101 book.
For $29.99, you can purchase a copy of my ebook, Testing Nutritional Status: The Ultimate Cheat Sheet, my complete system for managing your nutritional status using dietary analysis, a survey of just under 200 signs and symptoms, and a comprehensive guide to proper interpretation of labwork.
For $35, you can pre-order a complete bundle of my Vitamins and Minerals 101 book.
For $250-$1499.99, you can work one-on-one with me.
LabCorp results, drawn 9/9/2022.



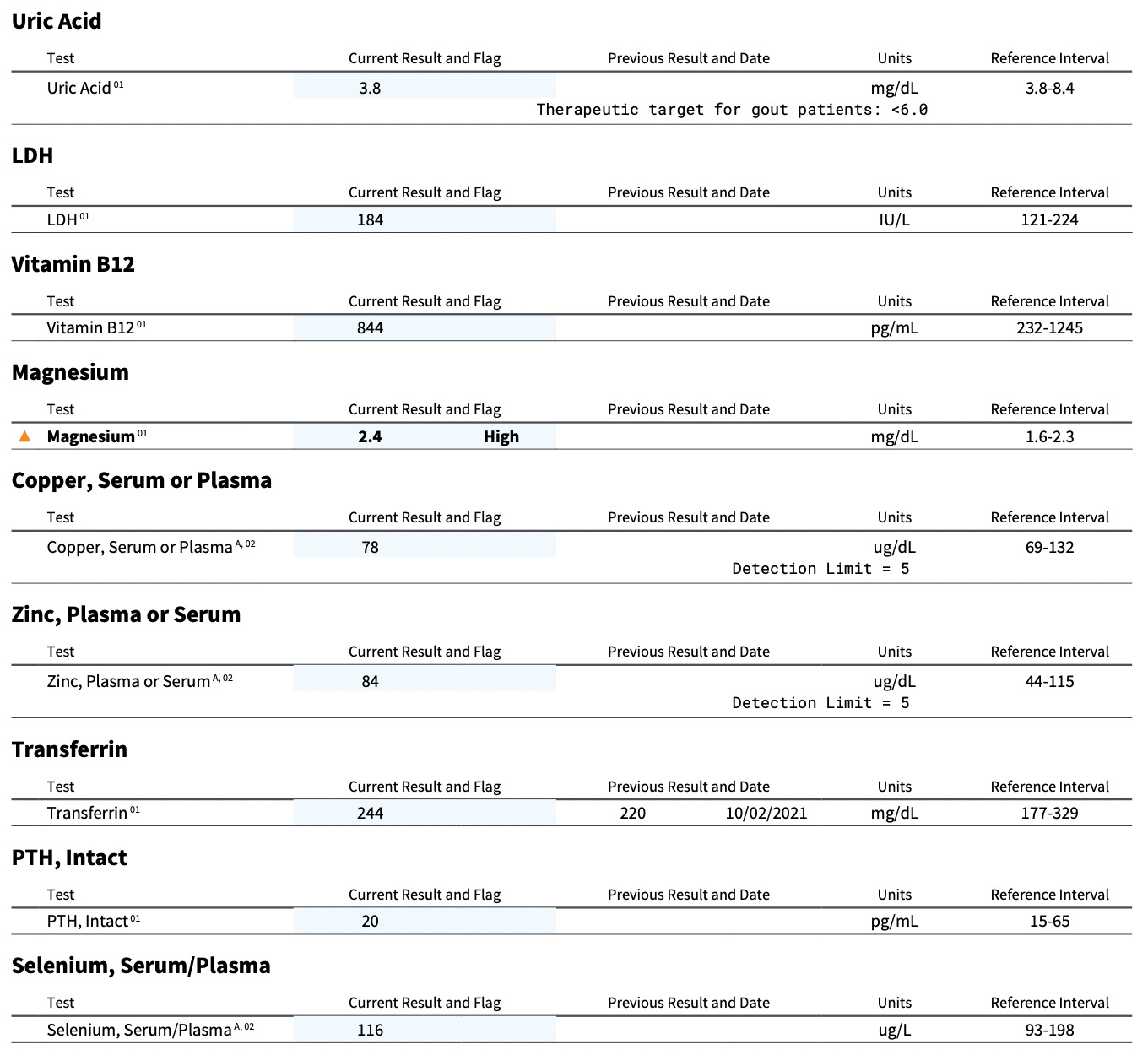
LabCorp results, drawn 10/7/2022


Genova ION, drawn May 23, 2017




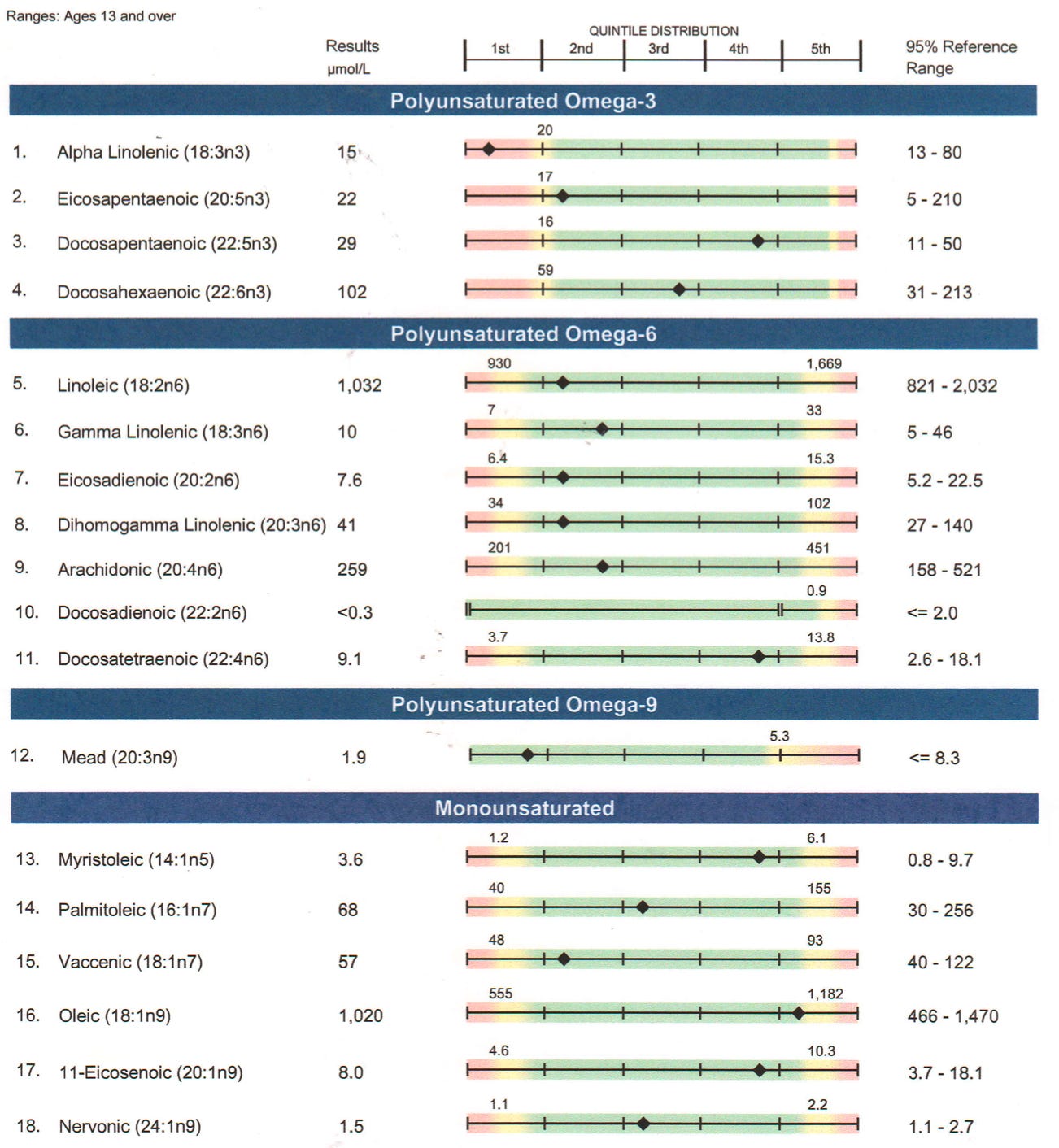



Genova ION, drawn September 27, 2022



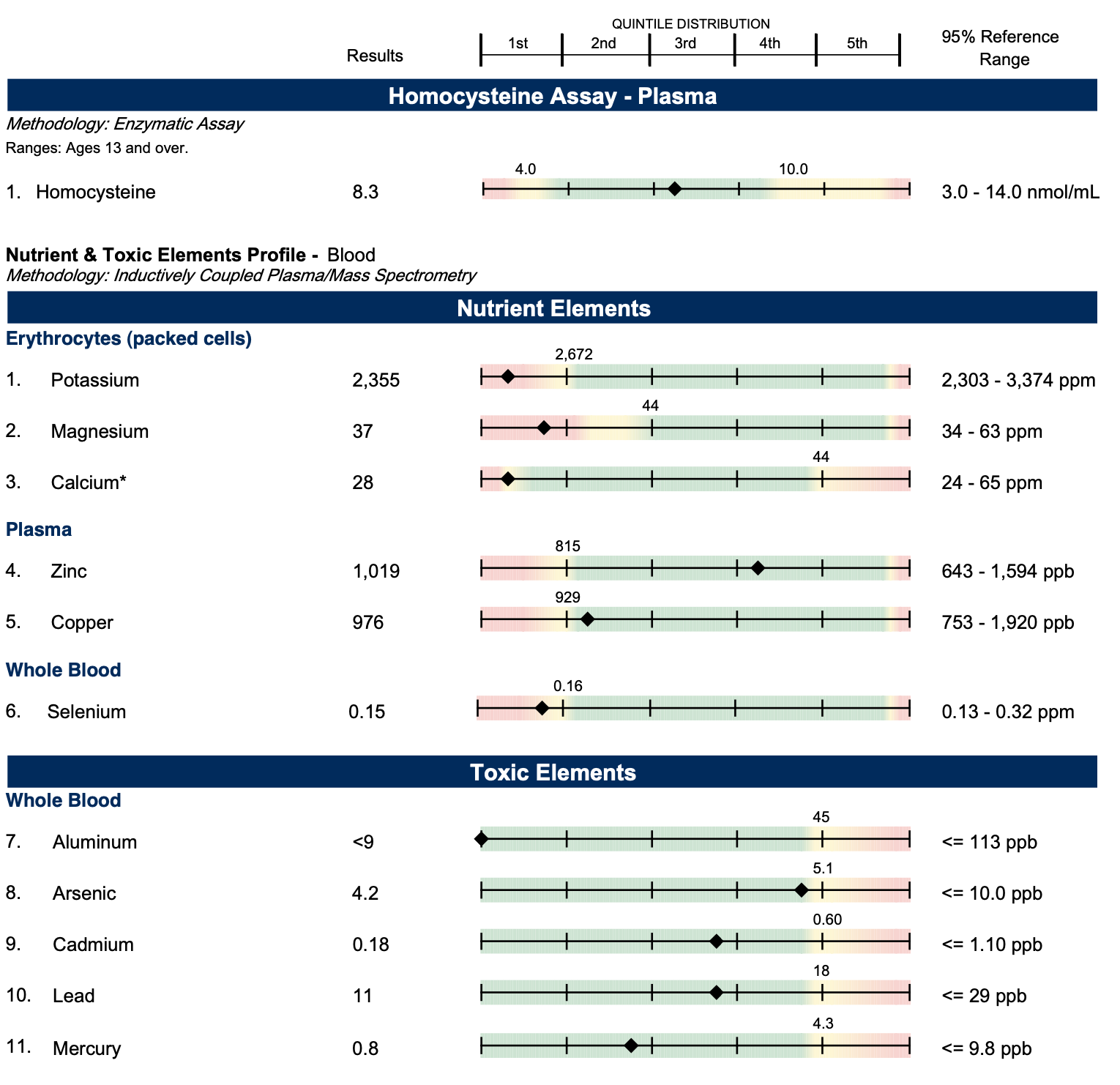


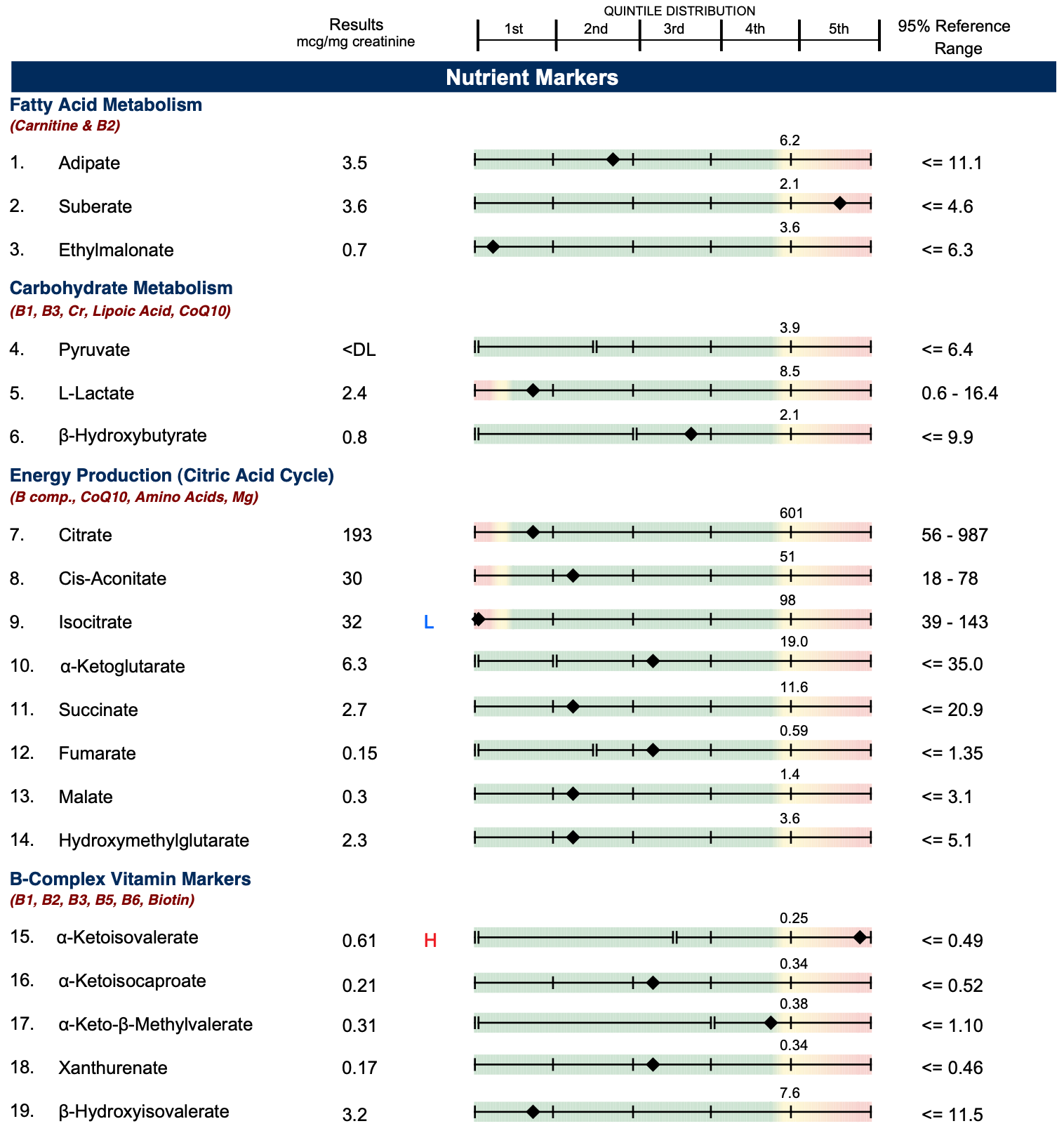

Genova Methylation Panel, drawn September 27, 2022

Van Coster, R. N., Fernhoff, P. M. & De Vivo, D. C. Pyruvate carboxylase deficiency: a benign variant with normal development. Pediatr. Res. 30, 1–4 (1991).
Hamilton, J., Rae, M. D., Logan, R. W. & Robinson, P. H. A case of benign pyruvate carboxylase deficiency with normal development. J. Inherit. Metab. Dis. 20, 401–403 (1997).
Wexler, I. D. et al. Molecular characterization of pyruvate carboxylase deficiency in two consanguineous families. Pediatr. Res. 43, 579–584 (1998).
Arnold, G. L., Griebel, M. L., Porterfield, M. & Brewster, M. Pyruvate carboxylase deficiency. Report of a case and additional evidence for the ‘mild’ phenotype. Clin. Pediatr. 40, 519–521 (2001).
Great Plains Organic Acids Test, drawn 12/20/2022


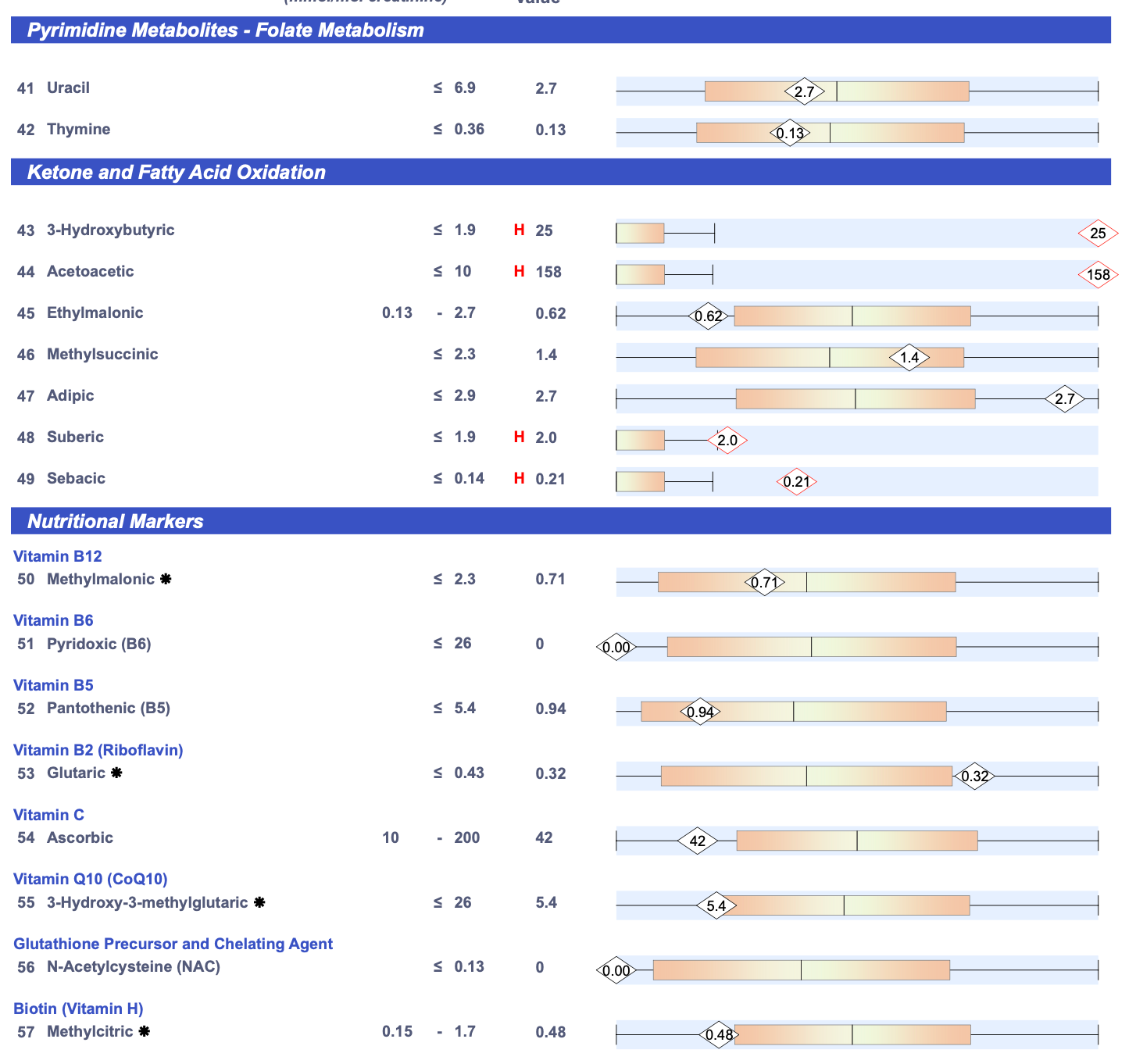

NutrEval, drawn on February 4, 2023




Quest Plasma Acylcarnitine Profile, drawn February 2, 2023




LabCorp, drawn February 6, 2023

LabCorp, drawn February 14, 2023

LabCorp, drawn February 6, 2023, 45 minutes after supplementation with 107 micrograms of biotin.

Went and got some chicken livers when I read the original article. Better now. Hadn't had them since forever.
I really enjoyed going through your test results. I see some similarities with my own, ie SAMe/SAH , biotin etc. I appreciate you being so open and posting, it's so helpful.