


Protecting Against Spike Protein Toxicity With Sulfur, Selenium, and Sunlight
Spike Protein Toxicity Part 2: Creating resilience with thioredoxin, glutathione, selenium, riboflavin, NADPH, and sunlight.

This is not medical advice. Please see the full disclaimer at the bottom.
In Part 1 of this series, The Spike Protein as a Pore-Forming Toxin, I outlined the evidence that the spike protein pokes holes in cell membranes at far lower concentrations than are found circulating after vaccination.
A study published just a few days later suggested this effect could be completely abolished in isolated cells with pharmaceutical inhibitors of heat shock protein 90 (HSP90).
HSP90 is a member of a broader family of proteins that faciliate the heat shock response. This response is induced by a variety of stresses, including infection and oxidative stress, but it is named after the most potent of these, heat stress. Heat shock proteins help protect cells from these stressors. The beneficial effects of using a sauna are mediated in part by increased heat shock proteins. On the other hand, extreme elevations of these proteins would be found in someone who died of heat shock. They are also thought to play a role in protecting cancer cells from various stresses including destruction by the immune system, and that is the main reason there are inhibitors available for research.
At first glance, this new paper would seem to suggest that HSP90 inhibitors are good candidates to try for protection against spike protein toxicity. However, so far none have been approved by the FDA so there are none available on the market.
Perhaps, then, we should look at natural ways to inhibit HSP90. However, clinical trials with HSP90 inhibitors for cancer have revealed gastrointestinal side effects like nausea and diarrhea; fatigue, muscle pain, and fainting; degeneration of the retina and visual disturbances such as blurred vision, flashes, and difficulty adjusting to the dark; and even liver failure and death.
Therefore, I believe a much better approach is to probe further into why HSP90 inhibition might be protecting against spike protein toxicity, and see if we can brainstorm nutrition and lifestyle interventions that would reap the benefits observed without all the harms that come with blanket inhibition of HSP90.
This brings us to sulfur, selenium, and sunlight.
What the HSP90 Paper Showed
The paper in question used human pulmonary microvascular endothelial cells and tested their ability to block the flow of electrical current. The method they used can detect disruption of the cell membrane or disruption of the junctions between cells, but they optimized it to test for the disruption of the junctions between the cells, which they refer to as “barrier dysfunction.”
They showed that whole spike protein as well as the S1 subunit caused barrier dysfunction. The S1 subunit was ten times more powerful than the whole spike protein. If they pre-treated the cells with HSP90 inhibitors four hours before adding spike protein, the effect was completely abolished. If they treated the cells with spike protein and then waited five hours to treat them with HSP90 inhibitors, the HSP90 inhibitors took ten hours to heal the cells and restore their behavior to normal.
To make sense of this, let us first take a step back and consider some background about the nature of these cells and their barriers.
Background: Endothelial Barriers and Their Permeability
Epithelium is a type of tissue that forms a protective layer on the outer and inner surfaces of structures throughout the body. It is joined by muscle tissue, nervous tissue, and connective tissue in making up the four types of tissue found in animals.
The endothelium is a specialized type of epithelium that forms the inner lining of blood vessels, through which blood flows. The endothelium is made up of endothelial cells. These cells have various types of junctions binding them together side to side, making up the endothelial barrier. The blood-brain barrier is one specific subtype of endothelial barrier. Behind them (that is, deeper inside the blood vessel wall), the endothelial cells make contact with muscle cells that control the relaxation and contraction of the vessel.
Endothelial cells sense what substances are flowing through the blood, how fast and smoothly the blood is flowing, and its pressure. They respond to these things, with the help of the muscle cells behind them, by coordinating the dilation or constriction of the blood vessel and the transport of substances small and large across the lining of the blood vessel wall in either direction, depending on what is needed.
The permeability of the endothelial barrier is highly regulated in a way that, in health, serves the needs of the many organs and systems to and from which the vessels are bringing blood. This barrier can become dysfunctionally hyperpermeable, however, and that plays a role in many diseases: asthma, pneumonia and acute respiratory distress as occurs in COVID, impaired gas exchange leading to hypoxia (low oxygen) or hypercapnia (excess carbon dioxide), arthritis, chronic bowel disease, cancer, stroke, edema (swelling), and impaired cognitve or motor functioning.
This picture from Wikipedia provides slightly more detail than we need:

Arteries and veins are the large vessels, functioning mainly to transport blood from one place to another rather than to transport stuff in and out of the blood. Arteries bring blood from the heart to other organs, while veins bring blood from other organs back to the heart.
Between them is the microcirculation consisting of the microvasculature or the microvessels. Arterioles bring blood into the capillaries. Capillaries exchange gases and other substances with the tissues with which they are enmeshed. Blood then flows through venules into the veins.
It is mainly the microcirculation that is permeable, befitting its role in transporting things in and out of the blood rather than merely moving the blood along in a certain direction. Permeability is highest in venules, least in arterioles, and intermediate in capillaries. The sensitivity to inflammatory stimuli that will increase permeability further follows the same pattern: greatest in venules, least in arterioles, intermediate in capillaries. Most transport of gases, electrolytes, and small molecules occurs in the capillaries. The transport of white blood cells occurs in the capillaries of the lungs, but otherwise usually occurs in venules. The transport of large molecules and fluid usually happens in venules.
The endothelial barrier is made up mainly of tight junctions and adherens junctions:
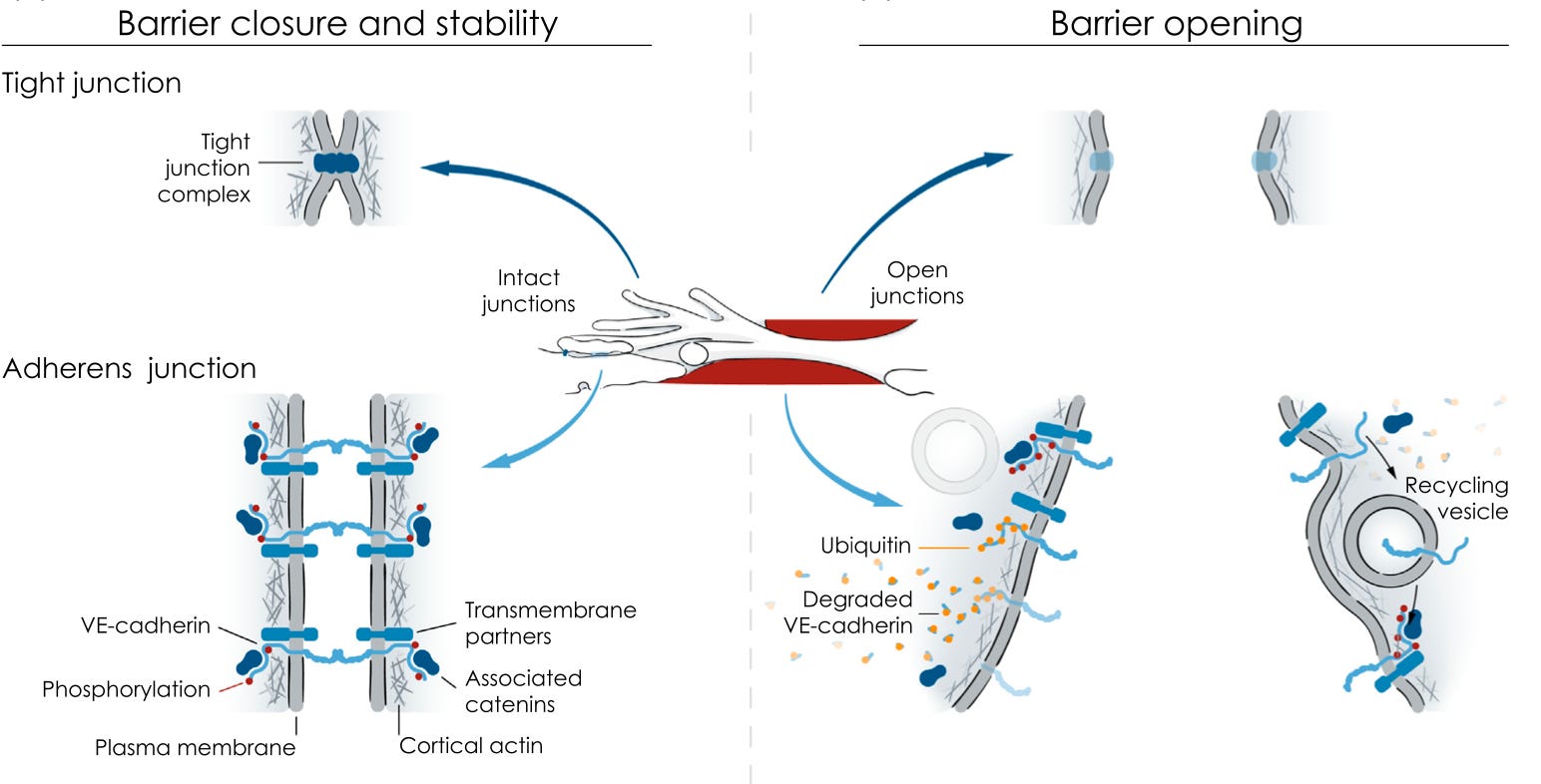
Tight junctions represent a more complete barrier and their quantity and tightness is the main determinant of how tight the barrier is overall. Adherens junctions appear to be more in the driver’s seat in responding to stimuli and opening up the barrier, although it is possible that their role in this is just better understood because it has been better researched. Tight junctions are usually found at the top of the endothelial cell facing the blood, while adherens junctions are usually found at the bottom of the cell facing the inside of the blood vessel wall.
The opening of the adherens junctions has been particularly well studied, and we will focus mostly on it for the remainder of this section.
Vascular endothelial cadherin (VE-cadherin) is a protein that stretches from inside one endothelial cell, across the membrane, across the space between cells, and into the inside of the neighboring cell. Inside each cell, VE-cadherin associates with other proteins known as catenins, including α-, β-, γ-, and p120-catenins. These either serve a regulatory role or help connect the cadherin-catenin complex to actin, a key protein of the cytoskeleton. The cytoskeleton is, simply put, the cell’s skeleton. It is like the human body’s skeleton only the proteins involved are different and it isn’t mineralized.
Various inflammatory stimuli — histamine, bradykinin, vascular endothelial growth factor, thrombin, platelet-activating factor, and TNF-α — open the barrier. A smaller collection of stimuli — beta-2 adrenergic agonists such as adrenaline, noradrenaline, and the bronchodilators used to treat asthma, and angiopoietin-1, a molecule involved in formation of blood vessels — close the barrier.
Background: The Role of Nitric Oxide and HSP90
The principle mechanism by which these stimuli open the barrier is through S-nitrosylation of the barrier proteins. This is a modification using nitric oxide and adding it to the sulfur found in the amino acid cysteine. Amino acids are building blocks of proteins, and when they are found bound within proteins they are referred to as residudes. The “S” in S-nitrosylation refers to the sulfur of the cysteine residue to which nitric oxide is added. S-nitrosylation is sometimes called S-nitrosation.
S-nitrosylation is completely distinct from the nitration of proteins, which is the addition mainly of peroxynitrite to tyrosine residues. Although it is possible nitration plays a regulatory role, protein nitration is usually associated with disease. By contrast, S-nitrosylation appears to be much more clearly and straightforwardly a reversible regulatory mechanism.
The figure from this paper shows the adherens junction in somewhat greater detail, with an emphasis on the role of S-nitrosylation:

Endothelial nitric oxide synthase (eNOS) is attached to the plasma membrane (the membrane surrounding the cell) at the site of a caveola, which comes from the Latin meaning “little cave.” Caveolae are cholesterol-rich sections of membrane with proteins known as caveolins that stabilize their invaginated “little cave”-like structures.
Although not shown in the figure above, eNOS is bound to the caveolin-1 protein and to heat shock protein 90 (HSP90). Caveolin-1 acts as an “off switch” and prevents nitric oxide synthesis.
As described in this paper, the various stimuli that lead to barrier opening as well as those that lead to the separate process of nitric oxide-mediated vasodilation generally lead to an increase in calcium ions inside the cell. Calcium then activates calmodulin, a regulatory protein (as its name implies, it “modulates” things in response to calcium). Calmodulin recruits additional HSP90 to eNOS. Calmodulin also binds to eNOS. HSP90 acts as a chaperone that helps calmodulin displace caveolin-1. This simultaneously allows eNOS to separate from the membrane and to shed its “off switch” and become activated.
Thus, HSP90 serves to enable calmodulin to activate eNOS in response to calcium.
Once activated, eNOS produces nitric oxide, which S-nitrosylates catenins and VE-cadherin, leading to their phosphorylation (another modification using the phsophate from ATP), and leading VE-cadherin to come into the cell, which breaks down the adherens junction.
With this background under our belts, let’s now turn to the new paper on spike protein toxicity and see if we can make better sense of it.
How Do HSP90 Inhibitors Protect Against Spike Protein Toxicity?
The most straightforward answer here is that inhibiting HSP90 prevents calmodulin from knocking caveolin-1 off of eNOS, which keeps it locked in the caveolae of the cell membrane and prevents it from making nitric oxide. This, then, prevents it from S-nitrosylating the adherens junction proteins and keeps the endothelial barrier intact.
This, however, does not explain why the spike protein would have activated calmodulin in the first place.
Let’s come back, then, to two things mentioned in part 1:
The spike protein allows charged ions to cross synthetic lipid bilayers that have no barrier proteins to mess with.
The spike protein is remarkably similar in its actions to pneumolysin, the pore-forming toxin made by Streptococcus pneumoniae, which is capable of causing pneumonia and respiratory distress on its own and which plays a role in Strep-induced myocarditis.
Pneumolysin, as well as a similar pore-forming toxin made by Listeria known as listeriolysin-O, disrupt the barrier of human lung microvascular endothelial cells just like the spike protein does. These authors showed that this is associated with the activation of eNOS and its shedding of HSP90 and caveolin-1. They suggested eNOS may have been activated by calcium entering through the pores that the toxins made in the cell membrane.1
Thus, I believe the most likely sequence of events is this:
The spike protein embeds itself in the cell membrane and makes it permeable to calcium.
Calcium comes in through spike-formed pores just as it would through a signal-activated ion channel under an inflammatory condition.
The calcium then activates nitric oxide signaling just like an inflammatory condition would.
Nitric oxide signaling then adds an entirely new form of permeability: it causes the adherens junction proteins to retract. On top of many “little doors” into the cells that the spike protein has made, nitric oxide signaling has now opened up many “huge doors” between the cells.
HSP90 inhibition blocks this second layer of permeability. It probably does nothing to fix the first layer of permeability — little holes made by spike proteins in the cell membranes — but it prevents this second layer of permeability that dramatically worsens the problem.
Blocking Nitric Oxide Release Is Not the Answer
Just as it would be rather foolish to use HSP90 inhibitors to solve this problem when they are associated with so many terrible side effects, including death, it would be rather foolish to try blocking nitric oxide release.
Nitric oxide plays two major roles that are likely to be protective in COVID, and probably in at least some forms of spike protein toxicity. It combines with glutathione to make nitrosoglutathione, which dilates the bronchioles and thus opens up the airways, and it acts by itself to dilate the blood vessels.
Nitric oxide is made from the amino acid arginine. My suspicion is that these roles of nitric oxide account for why L-arginine supplementation caused a seven-fold increase in the likelihood of respiratory improvement by day ten in a clinical trial of COVID patients with severe respiratory distress.
The question before us, then, is this:
Can we find a way to block the S-nitrosylation of adherens junction proteins while preserving the vasodilatory and bronchodilatory effects of nitric oxide?
Thioredoxin, Glutathione, Selenium, Riboflavin, and NADPH to the Rescue
The control of S-nitrosylation in the cell is a balance between three things:
Nitric oxide is always present, providing some baseline drive toward S-nitrosylation. When it increases, all things equal, more S-nitrosylation occurs.
Glutathione prevents S-nitrosylation by binding to nitric oxide to form S-nitrosoglutathione.
Thioredoxin, a selenium-dependent protein, denitrosylates proteins, removing nitric oxide from them.
To support the roles of glutathione and thioredoxin, there are two analogous enzymes that recycle them: glutathione reductase and thioredoxin reductase.
There will always be some baseline “tone” of S-nitrosylation in the cell, driven by the fact that there is always nitric oxide present and it is always counterbalanced by glutathione and thioredoxin.
However, anything that depletes glutathione or thioredoxin will increase S-nitrosylation, since even at baseline they are constantly keeping it in check.
If nitric oxide levels rise, the degree to which proteins become S-nitrosylated will depend on the relative rates of nitric oxide scavenging by glutathione, S-nitrosylation of proteins by unscavenged nitric oxide, and denitrosylation of those proteins by thioredoxin.
In this tug of war, nitric oxide will even inactivate thioredoxin by S-nitrosylating it to try to prolong its own signal. If thioredoxin can keep up, the active thioredoxins can denitrosylate the inactivated ones, freeing them up. Perhaps sometimes thioredoxin cannot keep up. This opens up the possibility that a large enough burst of nitric oxide could overhwelmingly S-nitrosylate the thioredoxin pool and push the system over the brink into a heavily S-nitrosylated state that it is much more difficult to recover from.
Therefore, the best protection against excessive S-nitrosylation is to maintain high levels of glutathione and thioredoxin, and a robust recycling system using their respective recycling enzymes.
However, would such an approach avoid compromising the bronchodilatory and vasodilatory roles of nitric oxide?
Are the Bronchodilatory and Vasodilatory Roles of Nitric Oxide at Risk?
We know that S-nitrosoglutathione is the endogenous bronchodilator. As such, glutathione is clearly essential to the ability of nitric oxide to open the airways, and thioredoxin-mediated protein denitrosylation is not a problem.
Since nitric oxide is thought to directly stimulate vasodilation on its own, however, whether this approach would hurt its effects on vasodilation deserves more comment.
Vasodilation is caused by the relaxation of the muscle cells that lie behind the endothelial cells within the blood vessel wall.
The most well established mechanism by which nitric oxide stimulates the relaxation of muscle cells within the vascular wall and thus the dilation of blood vessels is not dependent on S-nitrosylation of proteins. Rather, nitric oxide binds to an iron atom on the enzyme soluble guanylate cyclase, which makes it several hundred-fold more active, essentially switching it from “off” to “on.” This enzyme then catalyzes a cascade of other events leading to the relaxation of the muscle cells by inhibiting their contractile proteins.
Nevertheless, soluble guanylate cyclase can be S-nitrosylated, and that actually inhibits it. Glutathione prevents this.
In addition, there appears to be a second mechanism independent of this enzyme by which nitric oxide relaxes muscle cells, and it is directly dependent on glutathione.
The cytosol is the main compartment of the cell, and, in muscle cells, it is where the contractile proteins are. The concentration of calcium in the cytosol is the key signal that causes them to contract. Many signals can cause calcium to be released from storage into the cytosol (or to enter the cytosol from outside the cell). Reducing cytosolic calcium is the principal means of relaxing a muscle, and it requires pumping it into storage using a pump that depends on magnesium and ATP.
Nitric oxide combines with superoxide to make peroxynitrite. In the absence of glutathione, peroxynitrite can lead to damaging protein nitration. In the presence of glutathione, peroxynitrite oxidizes glutathione and the cysteine residues of proteins and makes them combine. This forms a modification known as S-glutathionylation. In the presence of glutathione, nitric oxide causes the S-glutathionylation of the calcium pump, which activates it. This leads to a reduction of cytosolic calcium, which then leads to muscle relaxation and dilation of the blood vessel.
In the absence of glutathione, nitric oxide is more likely to nitrate the calcium pump and contribute to its irreversible oxidative damage. This might contribute to the loss of blood vessel dilation known as “endothelial dysfunction” that shows up early in the course of cardiovascular disease. In the presence of glutathione, nitric oxide helps S-glutathionylate the pump, activating it, and contributing to the healthy relaxation of the muscle and dilation of the blood vessel.
In fact, glutathione has been shown to enhance nitric oxide-dependent vasodilation in isolated rat heart and when infused intravenously in humans.
Thus, supporting glutathione, thioredoxin, and their recycling enzymes is likely to protect against the ability of nitric oxide to cause excessive endothelial barrier permeability while preserving or even enhancing its ability to open up the airways and dilate the blood vessels.
Nutritional Support for Glutathione, Thioredoxin, and Their Recycling Enzymes
Glutathione and thioredoxin are both made from protein. Nutritional impacts on glutathione have been particularly well studied. Adequate glutathione synthesis requires one gram of protein per kilogram of bodyweight, which is roughly a half gram per pound of bodyweight and on the low end of what appears needed for healthy body composition.
Glutathione synthesis is influenced by four key factors:
In addition to adequate protein, the specific amino acids found in glutathione can boost its production. These are glutamate, cysteine, and glycine. Gluamate and cysteine combine to form glutamylcysteine, then glycine is added to form glutathione. Glutamate is rarely limiting because it is so abundant in the diet. Cysteine is usually limiting, but once it is supplemented glycine often is limiting. N-acetyl cysteine (NAC) is widely used to boost glutathione status. However, combining it with glycine makes a lot of sense, as was done in a recent study. Whey protein has an advantage over NAC in that its cysteine and glutamate are prepackaged as glutamylcysteine, which is more easily added to glycine to form glutathione.
Magnesium and ATP are both needed for its synthesis. A deficiency of magnesium, or a condition that lowers ATP — such as excessive fasting, overtraining and undereating, deficiencies of thyroid or adrenal hormones, common disorders of energy metabolism such as diabetes or rare genetic disorders — can compromise glutathione synthesis.
The ability to produce glutathione is signaled by a high ratio of insulin to glucagon, as occurs after a carbohydrate-rich meal. Since diabetics are either deficient in insulin or resistant to it, diabetes causes poor glutathione status. A euglycemic hyperinsulinemic clamp, which administers large amounts of insulin while keeping blood glucose stable, rescues the poor glutathione status of diabetes.
Oxidative stress signals the demand for glutathione and increases its synthesis. This can be mimicked by plant compounds known for their antioxidant activity, such as milk thistle or sulforaphane. A diet rich in fruits, vegetables, herbs, and spices provides a broad mix of such compounds.
Since thioredoxin is dependent on selenium, this raises the importance of selenium.
Glutathione and thioredoxin both must constantly be recycled by their respective recycling enzymes, glutathione reductase and thioredoxin reductase. Both of these are dependent on riboflavin, which they have bound as flavin adenine dinucleutide (FAD). They are also both dependent on niacin, which carries energy from glucose obtained through the pentose phosphate pathway in the form of NADPH.
In the pentose phosphate pathway, glucose becomes the source of NADPH, used for antioxidant defense, detoxification, recycling of nutrients like vitamin K and folate, and the anabolic synthesis of fatty acids, cholesterol, neurotransmitters, and nucleotides.
The pathway depends on an enzyme known as glucose 6-phosphate dehydrogenase. This enzyme is the target of a genetic deficiency, the most common such deficiency in the world, affecting about 8% of the population.
The pathway is enhanced by calcium and magnesium, dependent on iron, and critically dependent on thiamin.
On the other hand, there are two dietarily influenced conditions that can deplete NADPH: the conversion of carbohydrate to fat, and the conversion of sugars to sugar alcohols.
Under most circumstances, the conversion of carbohydrate to fat only totals one or two grams per day. However, a 70% carbohydrate diet where 60% of the carbohydrate comes from simple sugars will raise this toward ten grams per day. In premenopausal women, the conversion also reaches this high during the follicular phase, roughly the first two weeks of the menstrual cycle. These conditions might be marked by less NADPH available for glutathione and thioredoxin recycling.
The conversion of sugars to sugar alcohols happens when glucose or galactose levels exceed what can be handled by the normal routes of metabolism and glucose is converted to sorbitol or galactose is converted to galactitol.
In non-diabetics with fasting glucose ranging from 72 to 108 milligrams per deciliter (mg/dL) or 4-6 millimoles per liter (mmol/L), there does not seem to be any relationship between glucose and sorbitol levels. In diabetics, however, sorbitol in the cerebrospinal fluid starts rising when fasting blood glucose exceeds 126 mg/dL or 7 mmol/L, and red blood cell sorbitol starts rising when fasting blood glucose is above 97 mg/dL or 5.4 mmol/L.
Sorbitol, however, is going to accumulate postprandially and it is the peak glucose reached after a meal that should determine it. One study found that infusing glucose to 200 mg/dL (11.1 mmol/L) raises sorbitol in non-diabetics but not to the extent found in diabetics, and that infusing insulin in diabetics brings sorbitol down to normal levels when glucose gets under 150 mg/dL (8.3 mmol/L).
Higher sorbitol levels suggest NADPH is being depleted in the conversion of glucose to sorbitol and that less is available for the recycling of glutathione and thioredoxin.
Together, these suggest that avoiding diabetes is the overwhelmingly most important way to avoid sorbitol production, but that everyone should try to keep their peak glucose below 150 mg/dL. This is consistent with the cutoff of 140 mg/dL (7.7 mmol/L) often used to define healthy postprandial glucose.
Galactose is derived from lactose, the sugar in milk. Lactose, when digested, yields half glucose and half galactose. Galactose is overwhelmingly converted to glucose, but if too much is consumed at once, the excess can be converted to galactitol. One small study suggests that in the roughly half of people who show some galactitol in their blood after an oral galactose load, simply consuming the galactose every day for 15 days eliminates that effect. Based on this, starting a new milk habit might lower NADPH available for the recycling of glutathione and thioredoxin for no longer than two weeks.
Sunlight to the Rescue
Ultraviolet light denitrosylates proteins and releases the nitric oxide in free form. In a lab dish, two minutes under a UV lamp frees nearly all the nitric oxide that has bound to proteins. Blue and green light have a weaker effect.
Most likely, slight reddening of the skin (erythema) reflects increased blood flow consistent with the release of nitric oxide from proteins activating vasodilation, whereas anything resembling the inflammation of a sunburn, especially if it involves any puffiness or swelling (edema), reflects inflammatory activation of S-nitrosylation of proteins and barrier dysfunction. Therefore, sunlight is likely to have a beneficial effect up to the point of slight reddening but is likely to become detrimental by the time it reaches burning.
This is very unlikely to have any effect outside the skin. However, it may prove beneficial for any skin-related side effects of COVID or COVID vaccines, and it raises the possibility that internal phototherapy could be used for the healing of internal tissues.
My personal practice is to get at least 20 minutes of sunshine in the morning and at least 40 minutes of sunshine at peak UV (usually around 1:00 PM) whenever the UV index (using the Weather Channel app) is 5 or higher. During periods of the year where the UV index is consistently under 5 all week long, I replace the afternoon sunshine with a tanning bed once or twice a week. I find the morning sunshine helps my circardian rhythm and the afternoon sunshine prevents me from developing eczema.
Something similar could be useful for prevention or recovery from skin-related side effects of COVID and COVID vaccines.
Toward a Usable Protocol
In the political environment we face where it is dangerous to someone’s career to expose vaccine side effects, we are likely to be permanently left to a combination of mechanistic speculation and generalizing from the portion of COVID research that seems most relevant to overlapping effects of the vaccines.
Here, I will outline what I consider well supported mechanistic speculation for dealing specifically with the barrier dysfunction that results from the pore-forming toxicity of the spike protein. Somewhat arbitrarily, I would say for now that these considerations are most likely to be helpful in the full span of time (at least one month) leading up to the decision to get a COVID vaccine (for the life of me I can only see duress as a reason for this by now) and for a period of 3-6 months after the last dose, or throughout the duration of any side effects.
These suggestions would likely be helpful for COVID as well, from the point of contraction through at least the end of symptoms, including any that endure through any “long COVID” period. However, what is outlined in the COVID Guide (paid subscribers can access it for free here) takes precedence.
Make sure to consume at least one gram of protein per kilogram of bodyweight or at least a half gram of protein per pound of bodyweight.
Consider supplementing with 20-40 grams of whey protein, with anywhere from 500 mg each to 10 grams each of N-acetylcysteine and glycine, or anywhere from 500 mg to 5 grams of glutathione. My personal preference is for whey protein, supported by additional glutathione. I would stay toward 1500 mg of NAC or glutathione without a compelling reason to use higher doses, but consider what I’ve written here the top of the range to experiment with.2
NAC or glutathione might be tried topically for affected areas. (One of the studies cited herein used it on the inside of a hamster’s cheek at a concentration of 408 milligrams per liter.)
Keep diabetes well treated, and reverse type 2 diabetes if possible, and reverse any signs of emerging pre-diabetes.
Keep any thyroid, adrenal, or other metabolic disorder well treated.
Eat a diet that keeps postprandial blood sugar under 140 mg/dL (7.7 mmol/L). Within that constraint, eat freely of whole fruit and unrefined sweeteners, but use moderation and steer very far away from 42% of Calories coming from simple sugar. Within these constraints, any increase in carbohydrate is likely to improve glutathione status by increasing insulin.
Don’t start a new milk habit. If you use dairy products habitually, however, no need to stop.
Manage your vitamin and mineral status properly, with special emphasis on selenium, thiamin, niacin, riboflavin, iron, magnesium, and calcium. My Vitamins and Minerals 101 Cliff Notes are a great place to start (paid subscribers can access them for free here). I have a comprehensive system for managing nutritional status focused on lab work outlined in the Cheat Sheet, which is 50% off for Masterpass members, and paid subscribers get 50% off the Masterpass membership.
Specifically for any skin-related symptoms, get 40 minutes of sunshine per day during peak UV time, and when the UV index is under 5 try using a tanning bed at low doses. Always stay far away from a dose that causes a sunburn, and adjust longer or shorter duration depending on your skin tone. Consider using the D Minder app’s guidelines for vitamin D synthesis as a rule of thumb.
Based on the mechanisms discussed herein, people with uncontrolled diabetes or G6PDH deficiency might be at greater risk of vaccine side effects, as they are with COVID. The same would apply to untreated thyroid and adrenal disorders, and to any rare defects in energy metabolism.
Women might be at greater risk if vaccinated near the beginning of their menstrual cycle, since this would allow the highest ratio of spike protein to anti-spike antibodies to occur over the greatest length of the follicular phase.
The Bottom Line
It appears that the spike protein makes cell membranes more permeable to calcium. In the context of cells that line the inside of the blood vessels, this activates nitric oxide production and the S-nitrosylation of proteins involved in the barriers between cells, causing the barriers to become disrupted, thereby initiating a second layer of excess permeability focused on the gaps between cells.
Supporting glutathione and thioredoxin status is the best way to minimize this second layer of harm while preserving or even enhancing the beneficial effects of nitric oxide on dilating the blood vessels and airways. In the skin, exposure to sunshine without burning may help reverse some of the barrier dysfunction.
The “Toward a Usable Protocol” section outlines in more detail nutritional and lifestyle interventions that can support the roles of glutathione and thioredoxin.
Based on the mechanisms described here, people with uncontrolled diabetes, G6PDH deficiency, untreated thyroid or adrenal disorders, or genetic defects in energy metabolism should exercise extra caution around COVID and COVID vaccines. The beginning of the menstrual cycle might be the worst time for a woman to get a vaccine.
In the subtitle of this post I refer to “creating resilience” to spike protein toxicity. I believe what I have outlined here would be more powerful in reducing someone’s vulnerability to a vaccine side effect than in treating vaccine side effects. However, this is the proper starting place for thinking about treatment, because if the factors necessary for resilience are missing, the treatment will be that much more difficult.
This series is ongoing, and the outline of a practical protocol herein is preliminary. In future posts in this series, I will address how these principles apply to the gap junction proteins that protect against arrhythmia and the junctions in epithelial rather than endothelial barriers, and will address how to repair those barriers once damaged, in addition to addressing other mechanisms of spike protein toxicity and non-spike mechanisms of vaccine side effects.
Disclaimer
I am not a medical doctor and this is not medical advice. My goal is to empower you with information. I will not take a position on whether you should or should not get vaccinated. Please make this decision yourself, consulting sources you trust, including a caring health care professional.
Take Advantage of Your Masterpass Membership!
Ask a question in the Masterpass Forum, sign up for the next live Q&A, make sure you’re up to date on the premium content, check out the collections of ebooks and courses you have access to.
They also suggested that the loss of HSP90 and caveolin-1 from eNOS led it to make less nitric oxide and more superoxide. This switch is called “eNOS uncoupling” because eNOS is normally a dimer (two eNOS molecules bound together) bound by zinc-sulfur clusters, and when these clusters are oxidized or when eNOS is phosphorylated at a certain residue, it breaks down into monomers (single eNOS molecules), which make superoxide instead of nitric oxide. When this happens, it does not happen completely, so both nitric oxide and superoxide are made. They combine together to make peroxynitrite. These authors did take a portion of the caveolin-1 protein called the “scaffolding domain” and show that it helped reverse the eNOS uncoupling. However, they did not show that the barrier disruption was specifically due to the uncoupling. This is inconsistent with the fact that barrier disruption is known to be mediated by nitric oxide-induced S-nitrosylation. It is also inconsistent with the new study showing that inhibiting HSP90 prevents barrier disruption. If HSP90 prevented uncoupling and uncoupling caused barrier disruption, then inhibiting HSP90 would have made it worse.
This study used roughly 7.5 grams of glycine and almost 10 grams of N-acetylcysteine, which should be enough to synthesize about 18 grams of glutathione per day if used entirely for that purpose (unlikely of course). Dean Jones of Emory University in private communication suggested to me total glutathione synthesis per day is 10-20 grams per day, the top of which roughly lines up with this study. Therefore, I think it is reasonable to use what was shown in that study to be safe for NAC and glycine, or roughly half of what might be expected to be synthesized when using glutathione itself. Since oral doses that high of glutathione have not been tested in humans, I err on using the lower end of the range rather than the higher end.
Chris - Incredible report! Thanks so much. BTW, can't wait to read you book. Will it include a section with this info and treating covid intelligently (as opposed to mandated idiocy?)
Also, your interview with Jessica Rose was very informative. I can't count the number of times I have referred back to her Dec 16 article on the immune system pre and post vaccination...
https://jessicar.substack.com/p/the-bnt162b2-mrna-vaccine-against?s=r
Have you seen this article, which provides good evidence that UVA exposure reduces risk of COVID-19 in winter? The proposed mechanism is release of cutaneous NO.
Ultraviolet A radiation and COVID-19 deaths in the USA with replication studies in England and Italy.
Cherrie M, Clemens T, Colandrea C, Feng Z, Webb DJ, Weller RB, Dibben C.Br J Dermatol. 2021 Aug;185(2):363-370. doi: 10.1111/bjd.20093.