

D-lactate is commonly stated to be exclusively a microbial metabolite.
This is found in assumptions within the medical literature for decades even when it was long-known to be false.
You can watch the video, listen to the podcast, or keep reading.
This article is too long for email, so it’s best if you read it on the web site using the share button:
This is educational in nature and not medical or dietetic advice. See terms for additional and more complete disclaimers.
The microbial origin of D-lactate in humans was popularized in a low-quality 2018 paper in which subjects with small intestinal bacterial overgrowth had brain fog improve after taking a course of antibiotics and cutting out probiotics. Thirty percent of them (9 out of 30) had mildly elevated or high-normal urine D-lactate but there was no followup to show the D-lactate went down with the treatment, and thus no clear evidence it was of microbial origin. There was also no separation of the twin treatments of using antibiotics and cutting out probiotics and no untreated control group. Stories in Newsweek and Psychology Today spread the news that probiotics could cause brain fog by causing D-lactic acidosis, a conclusion that was premature in every conceivable respect.
While D-lactate is indeed made by bacteria, D-lactate is also inarguably and irrefutably produced by human enzymes.
D-lactate is produced endogenously from methylglyoxal, which can be made from glucose, threonine, or acetone, and is disposed of using the human enzyme D-lactate dehydrogenase, which depends on riboflavin and manganese, is embedded in the inner mitochondrial membrane, and almost certainly delivers the electrons to CoQ10, thereby feeding them directly into the respiratory chain. The D-lactate is thereby converted to L-pyruvate, which can then be burned for energy or converted to glucose.
Unlike L-lactate, which is excellent food for the brain, D-lactate is neurotoxic if it reaches levels in plasma that compete with those of L-lactate. High plasma levels of D-lactate lead to confusion, disorientation, slurred speech, difficulty coordinating movements, walking awkwardly, headache, hunger, irritability, aggression, hallucinations, and paranoia.
In this article, moreover, I will argue the following:
Microbial contribution to D-lactate in humans under normal circumstances is negligible.
I coin the term “the D-lactate shuttle” to describe a role for D-lactate that should eventually make its way into biochemistry textbooks alongside the malate-aspartate shuttle and the glycerol phosphate shuttle.
The D-lactate shuttle operates alongside these other shuttles to balance the priorities of conserving cytosolic NAD+, reducing cytosolic acidity, bypassing complex I, or generating ATP. It is uniquely useful as a shuttle when there is an absolute deficit of niacin or NAD(H).
D-lactate is an important contributor to gluconeogenesis that could account for up to 11% of it and rival an individual amino acid.
While D-lactate concentrations in human plasma are infinitesimal, when the downstream metabolism of D-lactate and L-lactate are blocked by genetic disorders, the concentrations of the two forms are similar in plasma. This contrasts wildly with the common claim that flux through D-lactate is “minuscule.” Most likely D-lactate is produced in considerable quantities in liver and kidney but is rarely secreted into plasma because doing so would risk neurotoxicity.
D-lactate should be taken seriously for its potential role in Parkinson’s and in neurological problems generally, for its role in diabetes, and for its extremely underappreciated roles in glycolysis, gluconeogenesis, and the respiratory chain.
Oxalate powerfully impairs D-lactate clearance, so D-lactate should be investigated as a potential link between oxalate and autism, and oxalate-lowering strategies should be seen as a way to improve D-lactate clearance and reduce its potential role in diabetes and neurological disorders.
See the sections on riboflavin, zinc, manganese, and glutathione in Testing Nutritional Status: The Ultimate Cheat Sheet, as well as Does CoQ10 Deserve a Spot on Your Longevity Plan? and the How to Detox Manganese guide for managing the relevant nutrients.
Look forward to more Masterclass with Masterjohn Energy Metabolism lessons on the D-lactate shuttle in the context of the related shuttles and the roles of L-lactate coming soon.
D-lactate is intimately intertwined with methylglyoxal, and our story begins at the beginning of the elucidation of the glycolytic pathway.
Methylglyoxal Was Originally Seen as Part of Glycolysis
In 1913, the German biochemist Carl Neuberg and, separately, the English biochemists Henry Drysdale Dakin and Harold Ward Dudley had demonstrated the existence of methylglyoxal and the glyoxalase enzymes that convert it to lactate. In the 1920s, Neuberg devoted his work to showing that this system was part of mainstream glycolysis.
“The formation of methylglyoxal,” Neuberg wrote, “is the essence of glycolysis.”
The German biochemist Gustav Georg Embden opposed this view, because in his hands glycolysis generated L-lactate exclusively, while methylglyoxal generated a mix of L- and D-lactate. By 1932, he had amassed a partial picture of glycolysis that “omits the methylglyoxal that was once accepted by Neuberg as the intermediary product in yeast fermentation as well as in glycolytic lactate formation.”
The German physiologist Otto Warburg worked out one of the steps of glycolysis, and the German biochemist Otto Meyerhoff confirmed much of Embden’s work after his death in 1933 and solidified what is today known as the Embden-Meyerhoff pathway of glycolysis.
In the 1930s through the 1950s, the details of the substrates and products of the glyoxalase system were worked out. During the 1970s-1990s, the glyoxalase enzymes were purified and characterized in detail.
Thornalley and Szent-Györgyi
Albert Szent-Györgyi won the 1937 Nobel Prize in Physiology or Medicine “for his discoveries in connection with the biological combustion process with special reference to vitamin C and the catalysis of fumaric acid.”
Szent-Györgyi was the first to isolate vitamin C, discovered the actin-myosin basis of muscle contraction, and discovered the role of fumarate in the Krebs cycle. He was also interested in ascribing a physiological role to methylglyoxal.
In 1963, Szent-Györgyi proposed that methylglyoxal was an inhibitor of growth, and that the glyoxalase system promoted growth by clearing away the methylglyoxal.
This fell out of favor due to the innumerable discoveries of other growth regulators, but I believe it is true, since methylglyoxal sequesters glutathione, and glutathione is needed for growth.
Thornalley wrote to Szent-Györgyi asking for guidance in picking up the torch of elucidating the physiological importance of methylglyoxal. Szent-Györgyi wrote back as follows:
I thank you for your kind lines from May 17th, and the reprints that reached me only now. It was very long ago that I was interested in keto-aldehydes and worked on them. Since then I forgot most of what I knew. So to my regret I am unable to advise you. Yours truly, Albert Szent-Györgyi, MD, PhD, NL
Thornalley framed this letter and put it on his office wall. He devoted his career to characterizing methylglyoxal as a toxic byproduct of glycolysis that causes diabetes through forming advanced glycation endproducts, with the physiological role of the glyoxalase system as a means of detoxifying methylglyoxal.
I believe this is true, yet as with oxidants (covered in my course on The Antioxidant System), I believe there are both physiological and pathological roles of methylglyoxal.
My Contribution on the Physiological Roles of Methylglyoxal
While my own original research on methylglyoxal focused on how dietary factors that impact glutathione levels can alter its accumulation, my doctoral dissertation laid out a synthesis of prior evidence to argue that methylglyoxal plays an important role in regulating redox balance during glycolysis, and serves a role in playing the balance between glycolysis and gluconeogenesis.
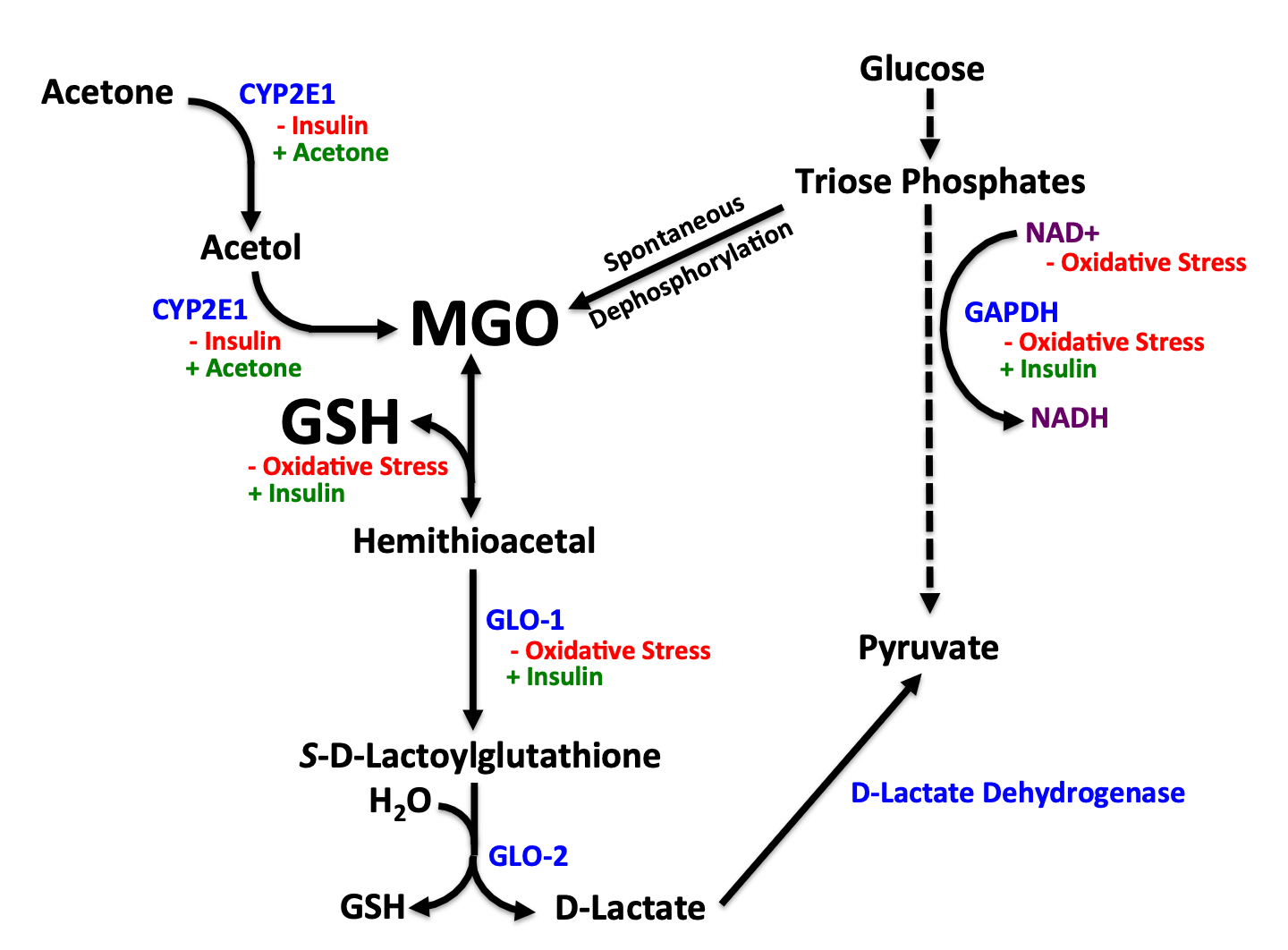
On a non-ketogenic diet, methyglyoxal comes primarily from glycolysis. Glucose is phosphorylated, becoming a hexose phosphate, meaning a phosphate of a 6-carbon sugar. It is split into two triose phosphates, glyceraldehyde 3-phosphate (GA3P) and dihydroxyacetone phosphate (DHAP). Their clearance through the rest of the glycolytic pathway is facilitated by NAD+ by the enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which is stimulated by insulin. Oxidative stress damages DNA, leading to the degradation of NAD+ for use during DNA repair. Though not covered in the figure, energy overload, carbs in excess of what can be burned, or impairments in complex I would raise the NADH/NAD+ ratio and also compromise GAPDH activity.
If NAD+ is inadequate for the GAPDH reaction, the triose phosphates spontaneously dephosphorylate to form methylglyoxal (MGO).
Under ketogenic conditions, methylglyoxal is primarily derived from acetone in a two-step process mediated by CYP2E1. This pathway is strengthened by acetone and suppressed by insulin.
The derivation from threonine is poorly understood and therefore not included in the figure. However, in vitro evidence suggests threonine generates methylglyoxal instead of glycine during its catabolism if coenzyme A (CoA) is limiting, and CoA would be expected to become more limiting under conditions of fatty acid oxidation.
Thus, under glycolytic conditions we can expect methylglyoxal to come from glycolysis, while under conditions dominated by fatty acid oxidation we can expect methylglyoxal to mainly come from acetone and threonine.
Methylglyoxal spontaneously reacts with glutathione to form a hemithioacetal adduct that is then acted on by glyoxalase-1 (GLO-1) to form S-D-lactoylglutathione and glyoxalase-2 (GLO-2) to form D-lactate. Glutathione and GLO-1 activity are both increased by insulin and hurt by oxidative stress. Both GLO-1 and GLO-2 are dependent on zinc.
D-lactate is the exclusive product of this pathway, and it is converted to L-pyruvate by D-lactate dehydrogenase.
Methylglyoxal rises in diabetes due to the loss of insulin signaling and the increase in oxidative stress, which acts to increase methylglyoxal formation and decrease its clearance. Methylglyoxal is the principle cause of advanced glycation endproducts (AGEs) in the human body. In diabetics, methylglyoxal and its corresponding AGEs are much higher than in healthy individuals. Animal experiments show that a single treatment with methylglyoxal can cause acute glucose intolerance while a month-long continuous treatment can induce advanced diabetes with pancreatic beta-cell failure.
But these AGEs don’t occur randomly. They are much more likely to occur on specific enzymes that carry out specific forms of regulation. In fact there are over 100,000 proteins, but proteomic analysis suggests methylglyoxal modifies about 500 of them. Glycolytic enzymes are especially targets. From among these, the single glycolytic enzyme known as fructose 1,6 bisphosphate aldolase (“aldolase”) stands out as the most often modified. Far more importantly, in 1978 research was published suggesting that the dihydroxyacetone phosphate that emerges from the aldolase enzyme starts generating methylglyoxal during its emergence. This extremely close proximity of glycolytically generated methylglyoxal to the active site of aldolase allows it to be uniquely potent in inhibiting this one enzyme.

Aldolase generates the triose phosphates that serve as precursors to methylglyoxal, so its inhibition of this enzyme acts as negative feedback against its own production.
It should also be seen as negative feedback against a rising NADH/NAD+ ratio. Aldolase generates the triose phosphates. NAD+ is needed to clear them away via GAPDH. In doing so, it converts to NADH. If the NADH/NAD+ ratio rises and GAPDH activity falls, continued aldolase activity will put more stress on the ratio by funneling substrate supply into GAPDH and encouraging it to go forward, generating more NADH, despite the scarcity of NAD+. Methylglyoxal’s inhibition of aldolase under this circumstance helps restrain continued worsening of the elevated NADH/NAD+ ratio.
As covered later, methylglyoxal also helps reduce cytosolic acidity and rewire electrons around complex I of the respiratory chain when complex I is overwhelmed.
My position on this differs from that of Berg, Tymoczko, Gatto, and Stryer, who in the 10th edition of Biochemistry (2023) refer to this as a “physiologically useless reaction.”
Meanwhile, methylglyoxal can be converted to pyruvate via D-lactate and be burned for energy in the mitochondrion.
When fatty acid oxidation predominates, acetone and possibly threonine become the primary sources of methylglyoxal. Fatty acids deliver 2-carbon units to the citric acid cycle without ever delivering 3-carbon units. The two carbons (green) are released as carbon dioxide, precluding the net synthesis of new glucose. The glycerol backbone of triglycerides (not shown) can contribute to some gluconeogenesis. Acetone can also lead to gluconeogenesis through D-lactate.
Gluconeogenesis requires aldolase. When methylglyoxal is generated from acetone or threonine, it is not close enough in proximity to inhibit aldolase, so gluconeogenesis proceeds.
Thus, glycolytic methylglyoxal acts as negative feedback on the step that increases cytosolic NAD+ demand and acts as a shunt to get around that step. This is a physiologically important way of reducing stress on the cytosolic NADH/NAD+ ratio.
Methylglyoxal from acetone and threonine serves as a gluconeogenic substrate.
Its contribution to diabetes likely represents runaway loss of control in these pathways with vicious cycles of excessive methylglyoxal formation, excessive impairment of specific enzymes, and higher rates of non-specific damage to a broad number of proteins.
A Short History of D-Lactate
Embden had very early on noted that methylglyoxal could generate D-lactate, but the principle papers identifying D-lactate as the exclusive product of the glyoxalase system were published in 1954 and 1973.
According to reviews, the clinical relevance of D-lactate was first noted in grain-fed cattle in 1965 and in a human with short-bowel syndrome in 1979.
However, two years prior to the first case of short-bowel syndrome, one case in 1977 was published looking for a genetic inborn error of metabolism as a cause of D-lactic acidosis. A four-year-old was evaluated for his severe mental retardation. He had high urinary excretion of D-lactate, and his plasma D-lactate (0.6 millimoles per liter) was almost as high as his L-lactate (1.7 millimoles per liter). He had very little lactate in his stool. His uric acid was always elevated, which they attributed to competition between D-lactate and urate for renal excretion. Two courses of antibiotics did nothing to reduce the D-lactate. In fact, the second course raised his D-lactate. Noticing structural similarities between D-lactate and threonine, they tried several threonine-loading experiments. The first one raised his D-lactate but the others failed. They published his case as an inborn error of metabolism causing D-lactic acidosis through an unknown mechanism. 42 years later, this puzzle would be solved.
Between 1979 and 2018, there had been 98 cases of D-lactic acidosis documented in humans, nearly all as a result of short bowel-syndrome, and this repeating finding reinforced the concept that D-lactate is a microbial metabolite.
The search for a D-lactate dehydrogenase was long befuddled and confused by the belief that it would resemble L-lactate dehydrogenase. Many papers referred to D-lactate dehydrogenase activity being observed, but either did not identify the enzyme, or attributed the activity to a non-specific mitochondrial enzyme.
Completely overlapping the identification of D-lactate as the exclusive enzymatic endproduct of the glyoxalase system, medical literature regularly made claims that humans do not make D-lactate.
For example, a 1989 paper stated that while dialysis fluid contains a mix of D- and L-lactate, “mammals produce only the L isomer from pyruvate by the enzyme L-lactate dehydrogenase (L-LDH) as D-LDH is found only in bacteria.”
This statement is confused on multiple levels. D-lactate is produced by glyoxalase-II, unrelated to lactate dehydrogenase, and the human D-lactate dehydrogenase does not make any form of lactate at all, but rather irreversibly converts D-lactate to L-pyruvate. The first point was known at the time; the second was not.
In 2002, a putative mitochondrial D-lactate dehydrogenase with two isoforms representing alternative splices of one gene was reported. The same year, an Italian group led by Lidia de Bari showed that D-lactate is effectively transported into mitochondria to be oxidized and possibly used primarily for gluconeogenesis. de Bari showed in 2013 that D-lactate dehydrogenase is a riboflavin-dependent enzyme that is embedded in the inner mitochondrial membrane, facing the the matrix, which is the inside of the mitochondria, and that it facilitates the use of D-lactate to generate ATP.
In 2019, a Dutch group and then later an Israeli group independently reported genetic cases of D-lactate dehydrogenase deficiency.
The Dutch paper was a reanalysis of the 1977 case of apparent genetic D-lactic acidosis. At age 40, he still had very high urinary D-lactate, and 0.7 millimoles per liter D-lactate in plasma. He also excreted D isomers of 2-hydroxyisovaleric acid and 2-hydroxyisocaproic acid, which are metabolites of branched-chain amino acids that are usually excreted in L-form when these pathways are impaired.
Antibiotics did nothing to lower D-lactate, showing again that it was not of microbial origin.
A second patient had West syndrome, consisting of infantile spasms and developmental regression, and had a similar excretion profile.
They found rare homozygous mutations in the putative D-lactate dehydrogenase gene identified in 2002. They deleted the corresponding gene in zebrafish and it made their D-lactate skyrocket. They installed the human wild-type gene into the zebrafish and it got rid of the D-lactate. However, installing the pathogenic variants found in their patients could not get rid of the D-lactate. They thus showed that the gene coded for an enzyme that metabolizes D-lactate and the mutations impair the enzyme’s ability to do that.
This report solidified the identity of this gene as the LDHD gene, coding for the human D-lactate dehydrogenase.
Notably their inconsistent threonine loading experiments in the 1977 paper could be explained by threonine converting to glycine or methylglyoxal depending on whether CoA is limiting.
And it is now clear that the first human clinical case of D-lactic acidosis was not short bowel syndrome but rather was genetic LDHD deficiency.
The Israeli paper then reported a family with gout that they attributed to mutations in the LDHD gene. D-lactate levels were considerably higher than in the Dutch paper, ranging from 1.52 to 4.7 millimoles per liter. They injected D-lactate into mice and showed it increased uric acid by 54%. They speculated that excess D-lactate is excreted into urine coupled to the reabsorption of uric acid back into the circulation.
This fits well with the 1977 finding that the first LDHD patient always had elevated uric acid.
A Chinese group in 2023 published a Nature Communications paper reporting the crystal structure of mammalian D-lactate dehydrogenase. They wanted to report the structure of the human enzyme, but it was not soluble in the system they used to manufacture it in E. coli. So they manufactured the mouse enzyme instead.
It contains riboflavin in the form of flavin adenine dinucleotide (FAD), which is bound tightly but not covalently. It is dependent on manganese for activity, with no ability of magnesium to substitute.
It is totally unrelated to L-lactate dehydrogenase, which depends on NAD+ and does not depend on FAD.
It works on D-2-hydroxyisovalerate and D-2-hydroxyisocaproate, also found elevated in the Dutch paper. It has higher affinity for them than for D-lactate. However, these substances were found in orders of magnitude lower concentration in the patients of the Dutch paper, showing that this enzyme primarily works on D-lactate due to its higher concentration.
All of the pathogenic mutations they tested made the enzyme more easily destroyed by heat, suggesting staying cool and avoiding sauna, fever, and excessive exercise would probably help mitigate the disorder. Half of the mutations hurt the binding of riboflavin to the enzyme, suggesting the disorders might be fully rescuable with high-dose riboflavin in those patients.
This year, in 2024, an Italian group managed to manufacture the soluble human enzyme in E. coli, allowing the study the Chinese group had originally set out and failed to do. They found that, in vitro, it donated electrons to CoQ10. In contrast to the plant version of the enzyme, it cannot donate electrons to cytochrome C. As had been shown previously for bacterial D-lactate dehydrogenases and as was suggested for the human enzyme in the 2013 de Bari paper, they found that it is strongly inhibited by oxalate. In fact, it is the most sensitive enzyme to oxalate inhibition on record.
Thus, dietary strategies to improve CoQ10 status or lower oxalate levels might also be helpful in cases of genetic deficiencies.
Is D-Lactate Production “Minuscule”?
According to the Nature Communications paper, D-lactate production from methylglyoxal is “minuscule.”
The associated citation implies that this pathway is minor because typical concentrations of D-lactate in human serum are 11 to 70 nanomoles per liter, whereas those of L-lactate are 1 to 2 millimoles per liter. I would argue the optimal range for resting L-lactate is 0.5-0.9 and the postprandial range can extend to 1.2, but we can use 1-2 for this purpose. Since a millimole is one million times a nanomole, the top of that L-lactate range is 28,571 times the top of that D-lactate range.
There is a huge problem with this reasoning.
First, you can never make a conclusion about the quantitative flux through a pathway based on concentrations alone. Concentrations represent a snapshot in an instant of time, whereas flux through the pathway is dynamic and can occur at different speeds. This is like looking at an aerial photograph of a major highway at one instant during the day and using the photo and the photo alone to quantify how many total cars got from point A to point B over the course of the day.
Second, it assumes their occurrence in serum is an equally important reflection of their occurrence in the body overall, but this may not be true if D-lactate is primarily produced and utilized in gluconeogenic tissues such as liver and kidney without ever being released into the general circulation.
In the Dutch case report of LDHD deficiency, plasma D-lactate was 0.7 millimoles per liter. In the Israeli case report of LDHD deficiency, plasma D-lactate ranged from 1.57 to 4.7 millimoles per liter. 4.7 millimoles per liter is over 67,142 times the previously cited top of the normal range, and 3-5 times the upper limit of normal for L-lactate. Clearly, then, in humans, the minuscule blood concentrations are driven by effective downstream metabolism of D-lactate, not of minuscule production.
To compare this to L-lactate, we cannot simply look at genetic deficiencies of L-lactate dehydrogenase, because this enzyme is involved in producing L-lactate as well as clearing it. Further, this enzyme comes in multiple isoforms with different propensities to form or oxidize L-lactate, and complete hereditary absence of the isoform that preferentially oxidizes L-lactate does not elevate L-lactate. The rate of clearance is governed primarily by pyruvate dehydrogenase.
In a 1996 paper by Saudubray and colleagues, pyruvate dehydrogenase deficiency was associated with L-lactate in the 2.4-4.6 millimole per liter range, with pyruvate mildly elevated to 0.21-0.45 instead of the 0.08-0.21 range. This yields a range of pyruvate + lactate of 2.61-5.05 millimoles per liter. Another case reported of a 7-year-old boy showed values within this range. The maximal L-lactate in pyruvate dehydrogenase deficiency is nearly identical to the maximal D-lactate in LDHD deficiency, and including the pyruvate to represent the totality of what is blocked by the mutation in the enzyme does not meaningfully change the comparison.
Thus, when the clearance of pyruvate, L-lactate, and D-lactate are blocked, the resulting concentrations of L-lactate and D-lactate in plasma are nearly identical.
In We Really Can Make Glucose From Fatty Acids After All! O Textbook, How Thy Biochemistry Hast Deceived Me!, I cited a 1979 paper that administered radiolabeled acetone to fasting subjects and found that its conversion to glucose could account for up to 11% of gluconeogenesis, which is comparable to an individual amino acid.
This is equal to production of 10 grams of D-lactate per day from acetone alone during fasting.
I have not seen gluconeogenesis rates from threonine quantified in humans, but in rats, 2.2-3.9% of glucose can come from threonine, which, extrapolated to humans, suggests that an additional 2-3.5 grams of glucose could come from threonine. Threonine has several pathways to glucose, but we can simplify this by saying the maximal flux through D-lactate during extended fasting could be as high as 13.5 grams per day.
This is very much at odds with Thornalley’s estimation using human red blood cells that 0.089% of glucose generates D-lactate. On 150 grams per day of net carbs, this would be 133 milligrams of D-lactate, and on 300 grams net carbs this would be 266 milligrams of D-lactate.
This discrepancy can be resolved if D-lactate is made primarily in the liver and kidney and not by red blood cells.
This is supported by the measured rates of uptake of these compounds from blood to liver. In rats over the course of 1.5 minutes, D-lactate is taken up 80% faster than L-lactate, but it is also sent back into the blood at a higher rate, such that the retention of L-lactate by the liver is 85% higher. In a similar but 2-hour long experiment in dogs, L-lactate was net retained in the liver at a 3.3-fold greater rate than D-lactate.
In bovine liver, maximal oxidation rates for L-lactate are 2.4-fold higher than for D-lactate.
This supports a model where the higher oxidation rate for L-lactate in liver drives a greater uptake of L-lactate from blood.
Consider these facts:
The net clearance of L-lactate from the blood into the liver 1.85-3.3-fold faster than the net clearance of D-lactate.
Serum concentrations of L-lactate are 28,571 times greater than D-lactate when their metabolism proceeds normally, but nearly identical when their metabolism is blocked.
If the liver were primarily taking D-lactate up from plasma, it would be completely impossible for it to generate a 28,571-fold differential favoring the L form in blood when it removes the L form faster than the D form.
This indicates that whereas the tissues that oxidize L-lactate are often different from the tissues that create it (for example, muscle and red blood cells release it, the liver oxidizes it), the tissues that oxidize D-lactate are the same as the tissues that create it (for example, liver and kidney make it, but it never leaves those tissues into the blood).
D-lactate is likely wired to avoid the general circulation so it doesn’t cause neurotoxicity. By contrast, if it is generated in large amounts in tissues such as the liver and kidney without leaving them, this poses no risk of neurotoxicity and, as we shall elaborate on below, can be very useful.
D-lactate production is not by any stretch of the imagination “minuscule” according to available data.
Is D-Lactate Usually Derived From Intestinal Bacteria?
In short bowel syndrome, surgical removal of large portions of the small intestine causes carbohydrate to mainly be fermented in the colon, with massive bacterial production of D-lactate that can raise concentrations in serum to as high as 20 millimoles per liter.
Clearly under those circumstances the gut bacteria can become the overwhelming source of D-lactate production.
But is the gut microbiome the source of D-lactate under normal circumstances?
One way to test this would be to look at D-lactate concentrations in germ-free rats or mice, but I cannot find such data.
Another would be to see if D-lactate is higher in the portal vein than in the general circulation. Since the gut empties into the portal vein before reaching the liver, the excess in the portal vein could hypothetically be due to microbial fermentation. One study in pigs suggests these values are nearly identical. A later paper from the same group presents the data more precisely and again the values are nearly identical.
The main conclusion from this is that there is no signal of excess D-lactate in the portal blood, and therefore no signal of a significant contribution of the microbiome.
However, to be more generous to the microbiome hypothesis, the data are equally consistent with the gut tissue itself releasing no D-lactate at all, unlike the other tissues that release it into general circulation. Since about half of the liver’s blood supply comes from the gastrointestinal tract, we could posit a maximum contribution from the microbiome to be 50% of D-lactate flux that arrives to the liver through the blood.
However, when we consider the previous argument that the liver has to be the primary source of its own D-lactate, then even 50% of the flux through the blood is negligible.
Thus, the gut bacteria cannot possibly be a major source of D-lactate under conditions other than short bowel syndrome or an equivalent and similar abnormality in the gut.
To show D-lactate coming from the microbiome, we want to see how much D-lactate falls in germ-free rats and mice and how much it falls in humans treated with antibiotics. In conditions where D-lactate is coming from the gut, it should also be higher in the concentration of the portal vein blood than in the general circulation.
Methylglyoxal is Clearly the Source of Increased D-Lactate in Diabetes
Diabetes is one of the most common afflictions in the modern world, and it both causes and is caused by increased methylglyoxal production and decreased clearance. In rats, diabetes approximately doubles D-lactate in plasma and liver, and this is almost certainly driven by increased flux through methylglyoxal.
Accordingly individuals with diabetes have higher levels of methylglyoxal in their blood.
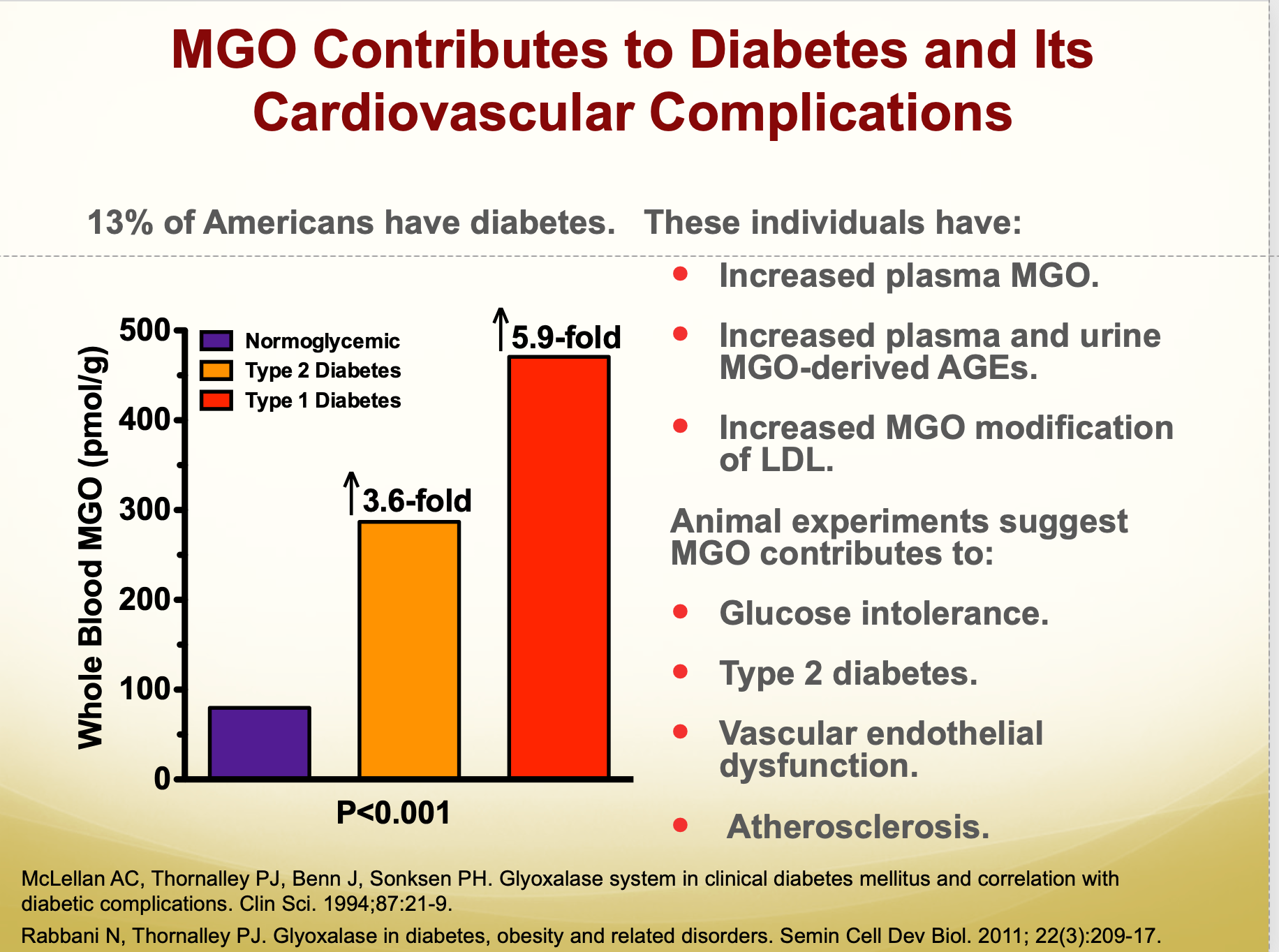
And higher concentrations of methylglyoxal-derived AGEs.
Thus, in pathogenic conditions involving increased D-lactate, the microbiome should not be the first suspect by any stretch of the imagination.
The demonstrated relevance of methylglyoxal to one of the most common pathological conditions afflicting modern society should be the first thing we look at if we find elevated levels of D-lactate.
What Is the Physiological Role of D-Lactate?
As covered in the beginning, I posited a physiological role of methylgyloxal in my 2012 dissertation that involves inhibiting glycolysis in a negative feedback loop under conditions of NAD+ limitation that otherwise favor glycolysis, and acting as a gluconeogenic substrate under gluconeogenic conditions.
We can now add some further nuances to this.
As background, mitochondria have two membranes. The area between the two membranes is the intermembrane space. The area inside the inner membrane is the matrix. Organic acids, ATP/ADP, NADH/NAD+, and ions can flow freely through voltage-gated anion channels in the outer membrane into the intermembrane space. The inner membrane has highly selective transporters that control the entry and exit of very specific substances into and out of the matrix.
In 2002, the de Bari group provided evidence that there are three inner membrane transporters that move D-lactate into the matrix, none of which transport L-lactate:
The putative D-lactate/H+ symporter moves D-lactate and hydrogen ions (acidity) into the matrix in the same direction.
The putative D-lactate/oxoacid antiporter moves D-lactate into the matrix coupled to the export of either pyruvate or oxaloacetate.
The putative D-lactate/malate antiporter moves D-lactate into the matrix coupled to the export of malate.
In that paper and in their 2013 paper, they showed the following:
D-lactate dehydrogenase is able to donate electrons to a synthetic electron acceptor added experimentally only when mitochondria are turned inside out, not when they are kept intact, which indicates that the enzyme is associated with the inner face of the inner mitochondrial membrane, facing the matrix.
It metabolizes D-lactate in a way that leads to ATP production and is not inhibited by the complex I toxin rotenone but is inhibited by antimycin, an inhibitor of complex III.
They did not explicitly address the question of what accepts the electrons from this reaction.
The 2023 Nature Communications paper authors stated explicitly that they would withhold any speculation on what the in vivo electron acceptor of this enzyme is.
The 2024 paper showed that, in vitro, it donates its electrons to CoQ10 but not to cytochrome C.
If we synthesize these three papers, this enzyme is almost certainly situated on the matrix side of the inner membrane with D-lactate binding on the matrix side, L-pyruvate leaving on the matrix side, and electrons flowing into the CoQ10 pool inside the membrane.
This figure from a CoQ10 review shows us what we should use as models to think about this.

The squiggly tadpole-looking thing is CoQ10. The enzymes shown in red are the matrix-facing membrane enzymes that deliver electrons to the CoQ10 pool. All three of these are FAD-containing enzymes that have their substrate-binding and product-releasing sites face the matrix and deliver electrons to the CoQ10 inside the membrane. They include enzymes involved in sulfur catabolism (SQR), proline metabolism (PRODH), and ETFDH, which accepts electrons from fatty acid oxidation, oxidation of branched-chain amino acids, lysine, and tryptophan, and from the scavenging of methyl groups from methylated metabolites of glycine.
These enzymes are similar to those shown in green, except the green ones have their substrate-binding and product-releasing sites facing the intermembrane space, which is toward the cytosol. Dihydroorotate dehydrogenase is involved in pyrimidine synthesis and uses riboflavin as flavin mononucleotide (FMN) instead of FAD, while mitochondrial glycerol 3-phosphate dehydrogenase depends on FAD like all the “red” enzymes.
Thus, all of the known riboflavin-dependent dehydrogenase enzymes embedded in the inner mitochondrial membrane either serve as a red or green enzyme in the figure, and donate their electrons to the CoQ10 pool.
While more experimental evidence is needed, until anything refutes my supposition, I think it is very clear that D-lactate dehydrogenase belongs in this figure as a “red” enzyme, with D-lactate being converted to L-pyruvate on the matrix side, and the electrons being dropped off at the CoQ10 pool.
From any of these sources, CoQ10 then delivers its electrons to complex III of the respiratory chain.
Introducing the D-Lactate Shuttle
The advantage of this pathway under glycolytic conditions would be that if the need for glycolysis exceeds the availability of cytosolic NAD+, glycolytic electrons can bypass the NAD+-requiring GAPDH reaction and can further bypass complex I and be donated instead to the CoQ10 pool, thereby removing stress on the NADH/NAD+ ratio caused by the backup at complex I.
I hereby dub this the D-lactate shuttle.
To my knowledge, I am the first to coin this term here in this article.
Biochemistry textbooks cite two such shuttles that bring electrons from the cytosolic NADH generated in glycolysis into the mitochondria. NADH cannot enter the mitochondria itself, so it is loaded onto malate in the malate-aspartate shuttle or onto glycerol 3-phosphate in the glycerol phosphate shuttle.
Technically D-lactate is not shuttling electrons from glycolytically generated NADH. Rather, it is shuttling the electrons that would have generated NADH in glycolysis but did not because they bypassed that step.
The malate-aspartate shuttle generates mitochondrial NADH that is oxidized at complex I.
The glycerol phosphate shuttle generates mitochondrial FADH2 that is donated directly to the CoQ10 pool.
The “D-lactate shuttle” generates mitochondrial FADH2 that is donated directly to the CoQ10 pool, just like the glycerol phosphate shuttle.
It carries electrons from glycolysis into the mitochondrial matrix on an organic acid, just like the malate-aspartate shuttle.
The glycerol phosphate shuttle shares in common with the D-lactate shuttle the funneling of electrons directly to the CoQ10 pool, but it does not transport the electrons by carrying an organic acid into the matrix.
Thus, the overlap in any two of the shuttles is similar to the overlap in any other two of the shuttles, so I consider all three comparable.
Thus, I believe it belongs in Biochemistry textbooks alongside these other two shuttles because it achieves very similar objectives in a very similar, yet distinct, manner.
If we remove methylglyoxal from the picture, this diagram summarizes how L-lactate production and the malate-aspartate and glycerol phosphate shuttles balance priorities of NAD+ demand, acidity, and respiratory chain capacity.

The first half of glycolysis is acidic because the ATP-mediated phosphorylations generate ADP + H+. The GAPDH reaction requires NAD+ and generates further acidity in the form of 2 NADH + H+.
Acidity impairs the two phosphorylation reactions, which would stop glycolysis, prevent the retention of glucose in the cell, and raise blood glucose. A high ratio of NADH to NAD+ impairs the NAD+-dependent GAPDH reaction. Thus, it is critical to rapidly neutralize the acidity and to remove the NADH.
The four phosphorylations in the second half of glycolysis neutralize all of the acidity produced in the first half, such that by the time we arrive at pyruvate glycolysis is net neutral.
As extensively documented by George Brooks and others, nearly all pyruvate is converted to L-lactate under ordinary circumstances, and there is some evidence that lactate dehydrogenase is often physically connected to GAPDH to facilitate the immediate conversion of NADH back to NAD+ so that the NAD+ needed for the GAPDH reaction continues in rapid supply.
L-lactate travels to the intermembrane space where the mitochondrial form of L-lactate dehydrogenase is physically connected to complex IV, and uses the energy released by complex IV to fuel the conversion of L-lactate back to pyruvate.
The intermembrane space is essentially continuous with the cytosol due to the lack of specificity of the transporters in the outer mitochondrial membrane. Thus, for all practical purposes the conversion of pyruvate to L-lactate in cytosol and the reversal of this reaction in the intermembrane space is net neutral toward cytosolic acidity, NADH, and NAD+. However, it facilitated the spatial distancing of NADH and acidity from the glycolytic enzymes, which is critical to keep them functioning at a high rate.
Both the malate-aspartate and the glycerol phosphate shuttle absorb one proton per molecule, bringing back the net alkalinity toward the cytosol that had occurred been forged by the production of lactate.
The distance of L-lactate oxidation from the glycolytic enzymes and its proximity to these shuttles means that L-lactate formation facilitated the net restoration of cytosolic NAD+ and the achievement of net alkaline glycolysis.
If complex I activity is able to keep up with the NADH produced from glycolysis, the NADH will use the malate-aspartate shuttle. This allows the production of mitochondrial NADH that is oxidized by complex I.
If complex I cannot keep up, the NADH will use the glycerol phosphate shuttle. This allows the production of mitochondrial FADH2 that is oxidized by the CoQ10 pool.
Skipping over complex I causes a 40% decline in ATP production. Therefore, it is not optimal. However, if complex I is overloaded, it generates reactive oxygen species that will cause broad negative feedback on all aspects of energy metabolism in order to prevent excessive damage to mitochondrial lipids and proteins. Therefore, skipping over complex I is necessary under certain conditions.
Now let’s fit methylglyoxal and D-lactate into this picture.

Methylglyoxal inhibits aldolase, which spares NAD+ by preventing the triose phosphates from driving GAPDH forward through substrate supply.
Forming mitochondrial L-pyruvate through methylglyoxal and D-lactate is accomplished with zero input from any form of niacin. No NADH, no NAD+, nothing.
D-lactate carries with it one proton per molecule into the mitochondrial matrix, which helps mitigate cytosolic acidity. However, the short-circuiting of the last half of glycolysis makes glycolysis net acidic. Thus, the putative D-lactate/H+ symporter mitigates the acidity but does not fully neutralize it.
The main advantage of the D-lactate shuttle compared to the glycerol phosphate shuttle is that it is much stronger in conserving cytosolic NAD+. It prevents its utilization by inhibiting aldolase, and it generates mitochondrial pyruvate without ever using NAD+.
The main disadvantage is that it is less potent in mopping up the acidity from the first half of glycolysis.
Both the glycerol phosphate shuttle and the D-lactate shuttle have the disadvantage that bypassing complex I leads to 40% less ATP.
In reality, these are all happening at the same time. Thus, the cell will turn the volume up or down on these three shuttles depending on its needs to conserve cytosolic NAD+, reduce cytosolic acidity, bypass complex I, or generate ATP.
Under gluconeogenic conditions, glycolysis is not moving forward. Methylglyoxal is derived from acetone and threonine instead of glycolysis, and it provides D-lactate for use in gluconeogenesis. The D-lactate enters the mitcohondrion in exchange for pyruvate, oxaloacetate, and malate entering the cytosol, all of which can be used for gluconeogenesis.
Does “Glyoxalase III” Spare the Brain?
Unbeknownst to me at the time, months before I finished my dissertation in 2012 a novel human glyoxalase unrelated to GLO-1 and GLO-2 was discovered and dubbed “glyoxalase III.”
The gene for the DJ-1 protein was discovered as an oncogene in mice in the 1990s and mutations in it were associated with early-onset Parkinson’s in the 2000s. It is now dubbed the PARK7 gene because of this.
Alongside those discoveries, in the 1990s, a gene in E. coli was dubbed GLO-3 for its ability to convert methylglyoxal to lactate without the use of glutathione or any other cofactor.
Realizing that the mammalian DJ-1 gene was related to the E. coli GLO-3, Korean researchers demonstrated cofactor-free glyoxalase activity for human DJ-1 protein in 2012.
In 2014, they showed that it converts methylglyoxal to L-lactate rather than D-lactate.
This is true for the human enzyme, but not the plant version. The authors noted that DJ-1 is highly expressed in brain, where LDHD expression is very low. Notably, plants don’t have brains, making the neurotoxicity irrelevant.
Indeed a tissue-based map of the human proteome published the next year found that LDHD is most highly expressed in liver and kidney, with considerable expression in pancreas, moderate expression in heart and skeletal muscle, and low expression in brain and many other tissues. PARK7 has high expression in most tissues, including brain. The gene for GLO-2, which generates D-lactate, has a somewhat similar pattern as LDHD. However, expression in brain is considerably lower than in liver and kidney but not minimal. Further, the gene for glyoxalase-1 (GLO1) is expressed fairly consistently across tissues, with higher expression in brain than kidney and liver.
Embden must have been correct that methylglyoxal generates a mix of L- and D-lactate.
Methylglyoxal makes a fast and spontaneous adduct with glutathione. The glutathionylated portion will generate D-lactate if the activity of GLO-1 and GLO-2 is high enough. The free portion is available to GLO-3 to make L-lactate. The cell can control the activities of these enzymes to bias the direction toward L-lactate, D-lactate, or — by maintaining a high ratio of GLO-1 to GLO-2 — keeping methylglyoxal sequestered as S-D-lactoylglutathione.
In the brain I believe we will have to dissect this at the cellular level to make sense of it.
Consider a few points:
Astrocytes feed neurons L-lactate as their primary fuel.
D-lactate dehydrogenase skips over complex I to feed electrons into the CoQ10 pool. This generates 40% less ATP than feeding electrons into complex I.
Blocking complex I with rotenone causes Parkinson-like dysfunction in animal models.
Mutations in PARK7, which block the conversion of methylglyoxal to L-lactate, cause early-onset Parkinson’s in humans.
D-lactic acidosis but not L-lactic acidosis causes neurotoxicity.
All this looks to me like D-lactate interferes with L-lactate in the brain by driving electrons into a 40% less efficient means of ATP production.
You want the versatility to use the D-lactate shuttle when complex I is overwhelmed and NAD+ is in short supply, but you don’t want complex I overwhelmed in the first place, you don’t want NAD+ in short supply, and you don’t want to force neurons to bypass complex I.
Some Practical Considerations
This article is primarily meant as biochemistry education and as a means of setting the record straight on D-lactate, so it is not meant for its practical utility.
However, some practical points can be made:
Quest has a plasma D-lactate test.
Home lactate meters are not isomer-specific. They are hard to compare to venous draws because they are a mix of venous, arterial, and capillary blood, along with interstitial fluid and fluid leaking out from damaged cells. Washing and swabbing the finger, generating large drops of blood, and wiping away the first one to three drops can reduce inaccuracies due to lactate on the surface of the skin or leaking from damaged tissue. If done properly, a large difference between a venous measurement of L-lactate and a finger prick with a home kit done at the same time could indicate the presence of significant D-lactate.
D-lactate is expected to be much higher in liver and kidney than blood, and under most circumstances blood measurements will not tell you what is going on inside cells.
The glyoxalase enzymes depend on zinc, insulin, and glutathione, and D-lactate dehydrogenase depends on riboflavin, manganese, and CoQ10. Testing Nutritional Status: The Ultimate Cheat Sheet has advice for optimizing the status of riboflavin, zinc, and glutathione. Does CoQ10 Deserve a Spot on Your Longevity Plan? has my tips for optimizing CoQ10 status. The easiest way to improve manganese status is to eat more plants. However, too much manganese causes CoQ10 deficiency, so optimizing manganese status is a careful balancing act. How to Detox Manganese covers ways to determine if you need to reduce manganese status and if so how to do it.
Since D-lactate dehydrogenase is the most sensitive enzyme to inhibition by oxalate on record, the possibility that oxalate contributes to autism should be suspected of involving impaired D-lactate clearance. Strategies to lower oxalate could include supplementing B6, avoiding vitamin C or collagen, reducing PUFA, consuming 300-400 milligrams of calcium per meal, reducing dietary oxalate, and perhaps optimizing status for biotin, folate, and B12.
The Bottom Line
Microbial contribution to D-lactate in humans under normal circumstances is negligible.
I coin the term “the D-lactate shuttle” herein to describe a role for D-lactate that should eventually make its way into biochemistry textbooks alongside the malate-aspartate shuttle and the glycerol phosphate shuttle.
The D-lactate shuttle operates alongside these other shuttles to balance the priorities of conserving cytosolic NAD+, reducing cytosolic acidity, bypassing complex I, or generating ATP. It is uniquely useful as a shuttle when there is an absolute deficit of niacin or NAD(H).
D-lactate is an important contributor to gluconeogenesis that could account for up to 11% of it and rival an individual amino acid.
While D-lactate concentrations in human plasma are infinitesimal, when the downstream metabolism of D-lactate and L-lactate are blocked by genetic disorders, the concentrations of the two forms are similar in plasma. This contrasts wildly with the common claim that flux through D-lactate is “minuscule.” Most likely D-lactate is produced in considerable quantities in liver and kidney but is rarely secreted into plasma because doing so would risk neurotoxicity.
D-lactate should be taken seriously for its potential role in Parkinson’s and in neurological problems generally, for its role in diabetes, and for its extremely underappreciated roles in glycolysis, gluconeogenesis, and the respiratory chain.
Oxalate powerfully impairs D-lactate clearance, so D-lactate should be investigated as a potential link between oxalate and autism, and oxalate-lowering strategies should be seen as a way to improve D-lactate clearance and reduce its potential role in diabetes and neurological disorders.
See the sections on riboflavin, zinc manganese, and glutathione in Testing Nutritional Status: The Ultimate Cheat Sheet, as well as Does CoQ10 Deserve a Spot on Your Longevity Plan? and the How to Detox Manganese guide for managing the relevant nutrients.
Look forward to more Masterclass with Masterjohn Energy Metabolism lessons on the D-lactate shuttle in the context of the related shuttles and the roles of L-lactate coming soon.
Brilliant! Love the "D-Lactate Shuttle" & the discussion adding other mitochondrial complexes to support this idea. The A.G.E linkage with methylglyoxal is particularly noteworthy as well as the resultant neurotoxicity. Including the oxalate effects and possible involvement with autism is also of great significance.
Will you recommend the use of calcium citrate since it is more effective? if yes is the dose per meal still 300mg to 400mg per meal?
Just came across these 2 research , Crystals in the Substantia Nigra ( https://pubmed.ncbi.nlm.nih.gov/31257859/ ) and Potentially Pathogenic Calcium Oxalate Dihydrate and Titanium Dioxide Crystals in the Alzheimer's Disease Entorhinal Cortex ( https://pubmed.ncbi.nlm.nih.gov/32804151/ ) . With these 2 papers, we should avoid using sunscreen containing titanium dioxide.