


SSRIs Can Cause Severe Mitochondrial Dysfunction
Installment eight in our series on understanding the truth about SSRIs.

In at least one out of 5,500 people – possibly at a much higher rate due to ignorance and underdiagnosis – sertraline (Zoloft) is such a potent mitochondrial poison that it can induce a devastating disease that has heretofore been thought to be a much rarer genetic disorder.
Adult-onset multiple acyl CoA dehydrogenase deficiency (MADD), also known as glutaric aciduria type II, is considered a rare genetic metabolic disorder with an incidence of one in 250,000, characterized by fat accumulation in skeletal muscle, exercise intolerance, and pain or weakness in the muscles.
Many cases may include other features, such as episodic vomiting, fatigue, difficult or rapid breathing, seizures, hypoglycemia, inflammation of the prostate, sensory neuropathy, and damage to the liver, heart, and skeletal muscle.
It turns out that sertraline (Zoloft) is 45 times more effective at inducing this otherwise genetic disorder than genetics themselves are.
In southeastern Sweden, there had been zero cases identified between 2004 and 2013.
Sertraline (Zoloft) was approved in Sweden in 2006 and in 2012 it became recommended as the first-line treatment for anxiety and depression. This increased the prescription rate by 48%. As of last year, 3.5% of people in Sweden were on sertraline.
Whereas zero cases of adult-onset MADD were diagnosed in southeastern Sweden between 2004 and 2013, nine cases were diagnosed between 2014 and 2024, but only two of them had a clear genetic basis.
In 2023, a North American group published a conference poster showing a woman who had a MADD-like biochemical profile on sertraline. The profile completely normalized with two measurements taken off sertraline. Going back on sertraline made the biochemical profile diagnostic for MADD again.
This prompted the neurology unit of the Linköping University Department of Biomedical and Clinical Sciences in southeastern Sweden to retrospectively analyze these cases for an environmental cause. All seven of the patients with no genetic explanation were on sertraline, and no other drug was taken by more than two people.
Whereas MADD is considered an extremely rare genetic disorder impacting one in 250,000, these cases imply that one in 5,500 sertraline users develops MADD. This is still rare, but it is no longer extremely rare. It implies that sertraline increases the risk of MADD 45-fold.
The publication of this paper rapidly led to similar retrospective analyses in western Sweden, North America, and Australia, bringing the total number of sertraline-induced MADD cases reported last year to thirty, with an additional two cases possibly attributable to the use of venlafaxine (Effexor) and duloxetine (Cymbalta), which are considered SNRIs rather than SSRIs because they inhibit the uptake of norepinephrine alongside that of serotonin.
That this one inquiry led to the immediate discovery of 30 cases of sertraline-induced late-onset MADD in just four of the world’s 197 countries is quite remarkable given that the total number of genetic cases ever reported worldwide is close to 600.
It is even more shocking when you see that, as we cover below, genetic MADD is cured with food-achievable doses of riboflavin, while sertraline-induced MADD seems to require going off sertraline to fix, and some people even then do not even recover but are left permanently or quasi-permanently with weakness, pain, neuropathy, and difficulty breathing.
Three case reports of genetic MADD published between 2012 and 2022 mentioned the patient was on sertraline without any further comment. Most MADD case reports do not even catalog the medications the patient is on at the time of symptom onset.
In severe cases, MADD and other disorders causing fat accumulation in muscle can lead to rhabdomyolysis, often but not always in response to exercise. This is a state of muscle breakdown where muscle proteins spill into the urine and make it darkly colored. Four case reports have been published on sertraline-induced rhabdomyolysis where it was not investigated whether it was driven by MADD. Given the other findings, these are likely severe cases of sertraline-induced MADD.
Given the general ignorance of this phenomenon and the fact that the question has not been asked in 193 out of 197 countries, these reports are surely just the tip of the iceberg.
This is educational in nature and not medical or dietetic advice. See terms for additional and more complete disclaimers.
This is the eigth installment in our series on the truth about serotonin and SSRIs.
Genetic Causes of MADD
MADD is considered a result of pathogenic mutations in the electron transferring flavoprotein dehydrogenase system, consisting of proteins encoded by the genes ETFA, ETFB, and ETFDH. This system is responsible for delivering electrons to the mitochondrial respiratory chain from the oxidation of all fatty acids; the amino acids lysine and tryptophan; the branched-chain amino acids leucine, isoleucine, and valine; and from dimethylglycine and sarcosine, which can be derived from glycine, choline, or betaine (trimethylglycine or TMG). The respiratory chain then uses these electrons to produce ATP, the primary energy currency of the cell. A backup in this system fundamentally impairs cellular energy production.
The system is dependent on riboflavin in the form of flavin adenine dinucleotide (FAD), so high-dose riboflavin supplementation is often helpful by improving the amount of FAD that can bind to, activate, stabilize the proteins involved and ensure their proper three-dimensional folding. Due to the importance of riboflavin, mutations that hurt riboflavin metabolism (in FLAD1, SLC25A32, SLC52A1, SLC52A2, and SLC52A3 genes) can also cause MADD.
While not often acknowledged as a cause of MADD, impairments in the respiratory chain from the point downstream of the ETF dehydrogenase system, from CoQ10 through complexes III and IV, could cause similar metabolic impairments.
Riboflavin deficiency, while it also has broader effects, is sufficient to cause an acquired form of MADD.
One of the major hallmarks is that an acylcarnitine profile shows many acylcarnitine species broadly elevated. These are carnitine-based detoxification products of intermediates in metabolic pathways that could not be completed. For example, derivatives of fatty acids or amino acids that are not fully oxidized will be detoxified using carnitine and will show up on the acylcarnitine profile.
The Centrality of Food-Level Riboflavin to Genetic MADD
Bizarrely, riboflavin status is often not even measured in this disorder, despite it universally being recognized to often be fixed by riboflavin supplementation. Instead, they just put people on high doses (10-150 milligrams per day in some cohorts, in others 300-400 milligrams or more) of riboflavin without testing whether riboflavin-rich foods or low-dose supplements would have worked.
Out of almost 110 cases at one center, a total of 15 people had their riboflavin tested prior to being treated with riboflavin, and they all had lower riboflavin status than healthy controls.
This raises the question of whether suboptimal riboflavin status is required to cause MADD in genetically susceptible individuals. If so, this contrasts wildly with the perception that high doses are required to fix a problem that could not be fixed with food or low-dose supplements.
Out of 48 patients with long-term followup in this cohort, the average dose used was 68 milligrams per day and the average followup was 10 months, and all 48 were “cured.”
Riboflavin generally cured them within two months. 30 of them stopped taking the riboflavin, but in 17 of them the disorder came back and went away when they resumed riboflavin. Some of them stayed on high doses and some of them transitioned to low doses or intermittent doses and none of this made a difference in the treatment outcome.
The return of the disorder after abandoning riboflavin was triggered by stressors such as physical exhaustion, catching a cold, drinking alcohol, or getting pregnant.
The patient in this case series who proved he only needed small doses of riboflavin to cure himself was one who initially cured himself in two months with 10 milligrams per day. Over the course of nine years, every time he had an attack of muscle weakness, he took five milligrams per day and it disappeared within a week.
Clearly he started from a very deficient position that needed a higher dose and duration to fix, then he progressed extremely slowly back across the threshold of deficiency and could pull himself out of it with a lower dose for a shorter period of time.
As I have covered in my abundant riboflavin materials, you can absorb 37 milligrams of riboflavin at a meal despite needs ranging from 1 to 6 milligrams per day. So these cases where people are cured with 150 milligrams are likely flushing high concentrations of riboflavin through the system to fix the deficiency quickly, then when the deficiency is fixed, the disorder disappears until riboflavin status sufficiently declines to the deficient state again.
Genetic MADD thus appears to be remarkably similar to MTHFR C677T in its riboflavin requirement, where bringing riboflavin from 1.6 milligrams per day to 3.2 milligrams a day fully resolves the function of the enzyme.
While the medical treatment of MADD seems largely incompetent in its failure to assess riboflavin status and use the minimal effective dose, the supplement industry is completely incompetent. If genetic cases of MADD only need 5 milligrams long-term, how is it even conceivable that the average person without a rare genetic disorder needs more? Yet it is nearly impossible to find a supplement of riboflavin less than 100 milligrams. Exceptions include a 36.5 milligram Thorne supplement or a 6.5-milligram liquid supplement, but even the Thorne supplement is quite high and not everyone’s digestive system will tolerate the glycerin in the liquid supplement.
In this cohort, recurrence and severity was not related to the specific gene mutations but was higher in people eating a vegetarian diet, which is generally low in riboflavin compared to an omnivorous diet.
This is because the best food for riboflavin is liver; the second-best foods are kidney, heart, and almonds; the third-best foods are red meat, cheese, eggs, salmon, mushrooms, seaweed, sesame, and wheat germ and bran; the fourth-best foods are milk and most other meats. Thus, you can design a vegetarian diet to be rich in riboflavin if you focus on almonds, mushrooms, seaweed, sesame, and whole wheat, but the average omnivore is going to get more by eating meat, eggs, and dairy food, and you have to eat organs to push your riboflavin up to the top of the range.
The one other risk factor for recurrence aside from vegetarianism was weakness in the muscles used for chewing. This is probably because these people had more difficulty eating food and therefore were more malnourished.
This is a rather shocking indication that the quantity of riboflavin in foods – most people get 1 milligram per day from food and you might hit 6 milligrams if you really design your diet around it – is more powerful a driver of MADD than genetics are.
That is, genetics load the gun and your diet pulls the trigger, with the trigger being very sensitive to the difference between 1 and 6 milligrams of riboflavin.
Unfortunately, riboflavin does not straightforwardly cure sertraline-induced MADD, where the operant variable is that the patients are poisoning their mitochondria with a pill their doctor gave them.
Sertraline-Induced MADD in Southeastern Sweden
In the cohort of sertraline-induced MADD in southeastern Sweden, nine patients presented with constant muscle weakness, usually in the legs more than the arms. Six of them had neck weakness, and in three of them their necks were so weak they physically couldn’t hold their head up.
Six had various manifestations of neuropathy, with numbness, pain, or sensory disturbances in the hands, feet, and lower legs. Three had cramps, two had episodic vomiting, and one became severely tired after fasting and was found to be suffering from acidosis.
All of them had accumulations of fat in their muscles. Two had fatty liver. One had low muscle CoQ10 content.
The authors state that none had altered respiratory chain activity, but they do not describe the details of which parts of the respiratory chain were tested nor do they show the data, so this has to be treated skeptically. For example they could have been testing something like oxygen consumption in response to malate, glutamate, pyruvate, or succinate, which would qualify as testing respiratory chain activity, but would shine no light on the nature of an impairment in the ETF dehydrogenase system.
None of them had classical signs of riboflavin deficiency. However, consistent with the nutritional neglect that broadly characterizes medicine across the board, none of them had their riboflavin tested.
Two of them had been on cholesterol-lowering statins, which made their condition worse. When they stopped the statins, they partially improved. This is straightforward since statins are well established mitochondrial toxins.
While several of them were taking folic acid, vitamin B12, vitamin D, proton pump inhibitors, thyroid meds, anti-convulsants, steroids, or ACE inhibitors, none of the vitamins were used by more than four people and none of the drugs were used by more than two people. None of them were taking drugs expected to cause myopathy as a side effect.
Two of them had a clear genetic causation to the MADD and the other seven were all on sertraline.
The onset of symptoms occurred between five months and eight years after starting sertraline. Three patients had case histories showing a dose response: one developed symptoms after increasing the dose from 50 to 75 milligrams and the other two after increasing the dose from 100 to 150 milligrams.
Three of them had been on Effexor, Paxil, or Lexapro prior to Zoloft without having developed muscle problems.
All patients received 150-300 milligrams per day of riboflavin, while two patients also received two grams per day of carnitine, and one also received 1050 milligrams per day of CoQ10. Four fully recovered, four got better, and one showed no improvement and currently suffers from trouble breathing, sensory disturbances, and pain and weakness in the muscles.
All of the patients showed normalization of the fat content of their muscles despite one remaining symptomatic.
Seven of the patients had their acylcarnitines go back up without symptoms returning, implying that the metabolic disorder was reactivating despite continued riboflavin treatment.
Once the retrospective analysis raised suspicion of sertraline, sertraline was discontinued. There were five patients who had elevated acylcarnitines at the time of sertraline discontinuation and in all five cases the acylcarnitines dramatically improved after stopping the sertraline:
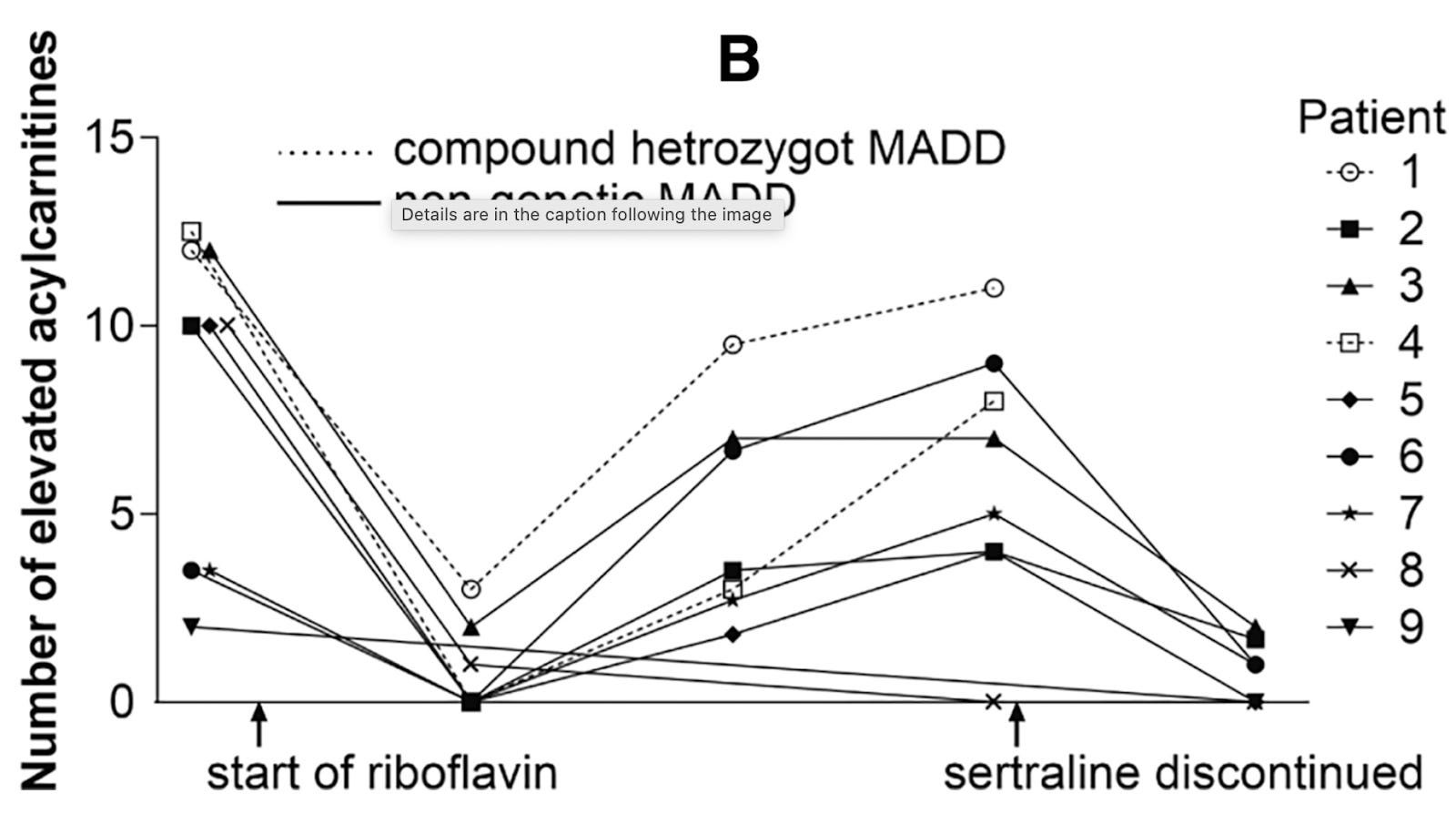
The return of elevated acylcarnitines while on riboflavin could imply a few things:
Sertraline was causing more and more damage over time, and eventually its damage outweighed the benefit of the riboflavin.
The riboflavin itself was a double-edged sword. On the one hand, it improved the ability to complete the oxidation of fatty acids. On the other hand, excessive long-term high-dose riboflavin-loading encouraged the body to increase the oxidation of fatty acids, thereby putting more load into the disrupted pathway.
While symptoms may not have returned, the return of the elevated acylcarnitines was probably a leading indicator that metabolic disruption was returning and that symptoms would have come in the future if the problem had remained unsolved.
The authors unfortunately did not explicitly comment on whether sertraline withdrawal made the one remaining symptomatic patient better, worse, or no different. They imply that the patient was still symptomatic at the time the report was written, indicating that sertraline had caused permanent or quasi-permanent damage, or that something beyond sertraline discontinuation was required to fix it.
It is also unclear whether “no clinical improvement” means this patient remained entirely in the same state or had actually gotten worse on the original riboflavin-based treatment.
Sertraline-Induced MADD in Western Sweden
The group from western Sweden found 17 individuals diagnosed with a lipid storage myopathy at the neuromuscular center in Gothenburg between 2015 and 2023. Two had a clear genetic diagnosis. Of the remaining 15, 12 agreed to further investigation, during which it was revealed that 11 of them were on sertraline at the time of diagnosis. These 11 were then further studied.
They all had weakness in the muscles closest to the center of the body, while most also had weakness in their extremities and about half had weakness in their neck. Most had muscle pain and sensory disturbances, while three had trouble swallowing.
Muscle biopsy showed fat accumulation in their muscles. They measured mitochondrial respiratory chain protein expression, but not enzymatic activity. There are dozens of proteins in the respiratory chain complexes, so they measured a representative protein from each complex. They also stained the tissues using antibodies to complexes I, II, and IV but not complex III. Overall these results suggested a profound defect in complex I, with mild and variable deficits of complexes II and IV and no evidence of a deficit in the more limited measurements of complex III.
Their mitochondria were abnormally small and numerous, with small numbers of them being abnormally big, and some appearing round with abnormal structures inside them.
They had no evidence of harm to mitochondrial DNA (mtDNA), though they had almost double the average number of mtDNA copies, suggesting their mitochondria were duplicating their DNA more than normal.
They then studied the total breadth of protein abundance (the “proteome”) of the muscles and found that nearly 2000 proteins were expressed differently in these patients compared to healthy controls. Proteins involved in helping the respiratory chain complexes assemble were upregulated, while the actual proteins that make up the respiratory chain (the subunits) tended to be downregulated. Complex I subunits were strongly downregulated, complexes II and IV more weakly so, and complexes III and V (V is ATP synthase) remained the same or were upregulated.
Most enzymes involved in fatty acid oxidation were upregulated, but ETFDH, which encodes ETF dehydrogenase and funnels electrons to the CoQ10 pool of the respiratory chain, was downregulated.
These last two changes alone would explain elevated acylcarnitines, since fatty acid oxidation being upregulated means there is more input into the pathway whereas ETFDH being downregulated means the pathway cannot run to completion. Fatty acids that start getting oxidized but can’t finish getting oxidized are the main driver of acylcarnitine accumulation.
Citrate synthase was upregulated, probably reflecting greater numbers of mitochondria. Together with doubled mtDNA content, this suggests increased mitochondrial biogenesis.
These patients had no evidence of genetic variants that could alter sertraline metabolism.
The authors did not measure riboflavin status, nor did they report treating these individuals with riboflavin or with sertraline withdrawal.
Sertraline-Induced MADD in North America
The North American group reported two cases of sertraline-associated MADD.
The first was a 26-year-old woman diagnosed with depression, profound fatigue, “psychogenic non-epileptic seizures,” subclinical hypothyroidism, and heart palpitations. Her acylcarnitine profile appeared as MADD.
She was counseled to take riboflavin and carnitine, avoid fasting, and to increase caloric intake when ill. This did not normalize her acylcarnitine profile.
However, going off sertraline did normalize her acylcarnitine profile, and going back on sertraline made it abnormal again:
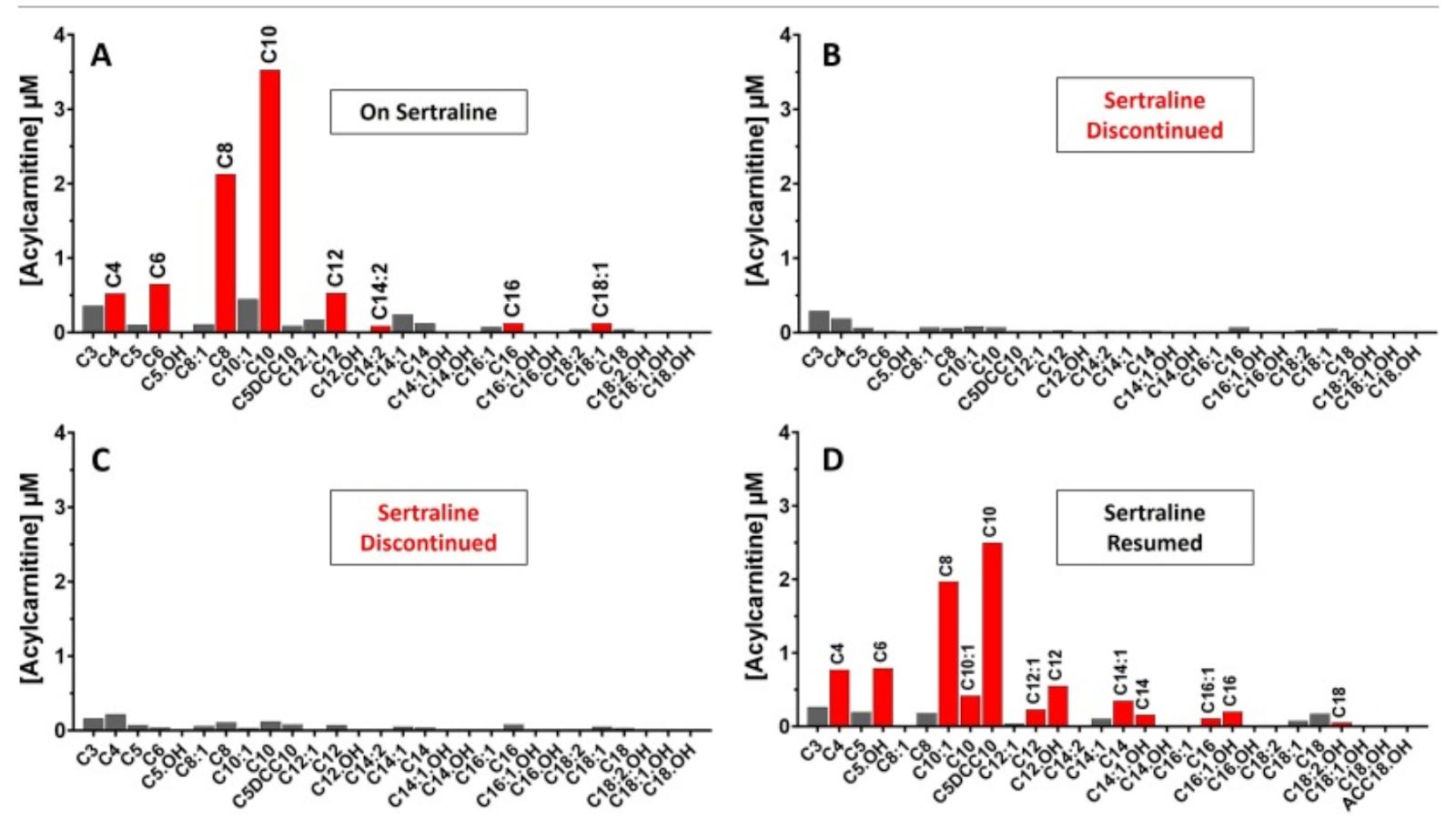
There were two measurements showing it normalized while off sertraline rather than just one, so this is especially compelling evidence that the sertraline was elevating the acylcarnitines and that random variation was not creating this as an illusion.
When combined with the data from southeastern Sweden, where five people all had their acylcarnitines normalize when they went off sertraline, we have two different angles – multiple measurements in one person, single measurements in multiple people – arguing against random variation and in favor of a real effect of sertraline.
After three months of staying on riboflavin and carnitine while eliminating sertraline, the first North American case reported “mild improvement in fatigue.”
The second case was a 61-year-old woman with chronic fatigue, exercise intolerance, weakness in the muscles closest to her trunk, and excessive tightness in her legs.
After a few months of these symptoms, she went to the ER after fainting and having difficulty swallowing and opening her mouth.
The ER found her in metabolic acidosis with rapidly increasing creatine kinase, a MADD-like acylcarnitine profile, and normal urinary organic acids.
Muscle biopsy showed fat accumulation and excessive proliferation of mitochondria with abnormal structures inside them.
Analysis of her respiratory chain activity showed isolated complex II deficiency.
Further investigation revealed she was on sertraline and had increased her dose of sertraline to 100 milligrams per day shortly before her ER visit. She was advised to discontinue it, and this made her creatine kinase rapidly return to normal.
She was further advised to take riboflavin and to moderate fat and protein.
After two years of taking riboflavin but avoiding sertraline, a repeat muscle biopsy showed her complex II activity had returned to normal and she no longer had fat accumulation.
Like the Swedish groups, the North American groups did not measure anyone’s riboflavin status.
It is very odd that they do not report anything about clinical improvement of the second case. Did her chronic fatigue, exercise intolerance, muscle weakness, and tightness improve as her muscle biopsy and respiratory chain activity normalized? Did she have any more episodes of fainting or difficulty swallowing or opening her mouth?
The silence on this question suggests she did not clinically improve, as was seen in one riboflavin-treated patient in the southeastern Swedish paper.
The first case had only “mild improvement” in fatigue, suggesting she was resistant to both riboflavin and sertraline discontinuation.
Sertraline-Induced MADD in Australia
In the Australian cohort, five of the eighteen cases of adult-onset MADD had traditional diagnostic criteria fulfilled for genetic MADD. Of the thirteen remaining, ten were on sertraline. Of the remaining three, one was on venlafaxine (Effexor) and one was on duloxetine (Cymbalta), both of which are SNRIs rather than SSRIs, which means they also inhibit the synaptic reuptake of norepinephrine alongside serotonin. Eight of the ten sertraline-associated cases had muscle biopsies performed, and all of them showed abnormal fat accumulation in the muscle. The patient on venlafaxine had a muscle biopsy and it showed mild inflammation but not fat accumulation.
Only eight of the eighteen adults in the combined adult-onset MADD cohort had riboflavin tested prior to starting riboflavin. Of them, half had low or borderline low riboflavin before starting supplementation. Of those who had normal riboflavin status, one was not prescribed riboflavin and the other three were. They did not respond to riboflavin at all. Among the four who had suboptimal status, three responded well to riboflavin treatment. While baseline riboflavin status was a critical determinant of the response to riboflavin, the genetic analysis had no ability to predict the response to riboflavin.
This again underscores that riboflavin “treatment” in MADD is overwhelmingly about optimizing riboflavin status and not on “treating” a highly specific genetic requirement for supraphysiological riboflavin concentrations.
All three patients who responded well to riboflavin had genetic MADD.
The anti-depressant associated MADD had a unique and different response to riboflavin. Riboflavin normalized the acylcarnitine profile in three and improved it in two, while failing to do anything in the others. The one patient taking venlafaxine had the acylcarnitine profile do nothing at first, but normalized six months after starting riboflavin. All of the patients whose acylcarnitine profile normalized or improved after riboflavin supplementation had them subsequently worsen several months later.
This is exactly what was observed in the southeastern Swedish cohort: riboflavin initially improved the acylcarnitine profile, then it worsened. The only difference is the Australian study did not take the patients off sertraline, so it was not able to show the renormalization of the profile after discontinuing the drug.
Of the four adult patients with suboptimal riboflavin status, three were on sertraline. Riboflavin induced complete recovery only in the one patient who was not on sertraline. In the three on sertraline, one had no response and the other two appeared to improve biochemically at first but then reverted back to their initial biochemical profile without improving clinically.
This is inconsistent with the tendency to improve clinically in the southeastern Swedish cohort and it emphasizes that there is a subset of people with sertraline-induced MADD who do not get better on riboflavin.
Unfortunately, the Australian paper did not report the doses of riboflavin used, and left us with the cryptic statement that they are now “trialing higher dose riboflavin in these patients” to see if it can indeed overcome the effect of sertraline. They speculated without any evidence that sertraline may interfere with the binding of FAD to ETF or ETFDH and therefore it may respond to supraphysiological riboflavin doses.
It is bizarre that they are not “trialling sertraline withdrawal” as the Australian medical establishment, unlike the North American or Swedish establishments, evidently consider it important to maintain their patients on the mitochondrial toxin they were originally prescribed.
The Bottom Line
The signal for sertraline here is very powerful. Other SSRIs and the SNRIs do not seem compellingly associated with MADD. The two Australian cases on SNRIs are counterbalanced by the three Swedish cases where other drugs used prior to sertraline did not cause MADD in the same individuals in whom sertraline did cause MADD. The North American case where two measurements off sertraline produced a fully normalized acylcarnitine profile in one individual while going back on sertraline caused it to resume its MADD-like state alongside five Swedish cases where going off sertraline all made an abnormal acylcarnitine profile normalize offers clearer evidence that this is causation rather than a simple correlation.
The fact that sertraline-induced MADD is 45 times more common than genetic MADD, that it is harder to fix, and that 30 cases were found almost immediately in four out of 197 countries while only about 600 total genetic cases worldwide have ever been reported suggests this is the tip of the iceberg and that this is a rare but powerful and dangerous effect of sertraline.
Indeed, how many people with fatigue, weakness, and muscle pain even get an acylcarnitine profile tested?
If this only occurs in one in 5500 people, or even if it is 100 times more common than this and it is one out of 55 people, this still implies it is a genetic idiosyncrasy creating a predisposition for it to occur. Within some relatively uncommon genetic constraints, however, it becomes a predictable dose-dependent effect of sertraline.
Several heterozygous mutations in the ETFDH system, riboflavin transport, or fatty acid oxidation had been potentially tied to some of these cases, but the defining feature of sertraline was that there was no basis for a classical genetic diagnosis involving two pathogenic mutations in the same enzyme, and even the Australian paper which cast the broadest net for genetic investigation could only find a single heterozygous mutation in three out of ten cases.
That doesn’t mean genetics aren’t involved. It suggests instead that a much broader net needs to be cast to find the relevant genetic predisposition.
Not a single one of these papers mentioned hypoxia or the sigma-1 receptor, showing that pharmacology, psychiatry, and the specialists in inborn errors of metabolism are fully united in completely misunderstanding the role of serotonin in the body and the intracellular impacts of SSRIs.
None of these cases will ever be solved until these disciplines exit the intellectual stranglehold in which SSRIs are seen as “psychiatric drugs” rather than “mitochondrial drugs.”
The question arises whether this can be explained by any of the known effects of sertraline.
The increased mitochondrial biogenesis implied in the western Swedish paper is consistent with the ability of extracellular serotonin to activate this process, though it could also be a secondary compensation for poorly functioning mitochondria.
The observations about the respiratory chain patterns in the western Swedish and North American papers seem to conflict with one another or imply clinical heterogeneity, but they cannot be directly compared due to fundamental differences in methodology. Nevertheless, they are both consistent with variations of the hypoxia response, which is centrally impacted by SSRIs.
Sertraline stands out as a powerful sigma-1 activator, a close second to fluvoxamine. Unfortunately, no studies have directly compared the rate of sertraline and fluvoxamine uptake into cells. It could be that while fluvoxamine is a more powerful activator of the sigma-1 receptor, sertraline enters cells much better than fluvoxamine and that this is enough to more than make up for the gap.
In general, SSRIs that strongly activate the sigma-1 receptor are expected to be better for mitochondrial function than those that activate it poorly or trivially.
However, there could be a genetic mutation found in these cases that fundamentally changes the importance of chronic sigma-1 activation. In fact, it could even alter the way sertraline binds to it without altering the way fluvoxamine binds to it. Since no one investigating this appreciates the importance of sigma-1 activation to sertraline’s impact on mitochondrial function, no one has looked at it.
Alternatively, SSRIs differ in how they structurally interact with the serotonin transporter, so there could be a mutation that makes sertraline especially potent in blocking the entry of serotonin into the mitochondria where it is needed to synthesize melatonin. Or perhaps it even makes it less likely for sertraline to dissociate from the extracellular serotonin transporter and therefore enter the cell in the first place.
It is not clear to what degree the elevated acylcarnitines reflect the centrally causal event in metabolic dysfunction or reflect impaired mitochondrial respiration as centrally causal. As noted in the beginning, impairments in the respiratory chain can elevate acylcarnitines. While the western Swedish paper also found elevated fatty acid oxidation and suppressed ETF dehydrogenase, it can’t be ruled out that this is secondary to hypoxia response activation, which can have variable impacts on fatty acid oxidation depending on the intensity and duration of hypoxia.
Thus, there is much more to learn about this, but it appears specifically tied to sertraline, clearly indicated by broadly elevated acylcarnitines, and sometimes but not always responsive to riboflavin supplementation and sertraline discontinuation.
Sertraline discontinuation can be complicated by the fact that SSRI withdrawal can cause mitochondrial dysfunction. However, I suspect that this would not occur in the case of sertraline-induced MADD. Neither of my clients who suffered catastrophic effects from SSRI withdrawal had anything that looked like this phenomenon, despite one who was on sertraline for years, and while clinical improvement was reported for some but not all sertraline-induced MADD patients, none of the papers reported any problems during withdrawal.
The ideal way to address riboflavin would be to first measure blood levels. Especially if they are low, focus should be placed on getting more riboflavin from food. A “riboflavin flush” with a high dose for two weeks might be valuable as a way to fix the deficiency much faster, but it should be followed by going back to food-level riboflavin. Supplemental riboflavin can be titrated up from 6.5 milligrams to 37 milligrams to 100 milligrams using liquid riboflavin, the Thorne product, and a standard 100-milligram product. You can also take a 100-milligram capsule and weigh it out into four 25-milligram capsules using empty capsules and a milligram scale to give you more control over the dose. In such a titration you want to slowly find the minimum dose that is needed to produce the maximally effective result. Going higher than this should be safe in most cases but should only be done if increasing the dose continues to generate big dividends, or if lower doses do nothing and you are temporarily exploring whether higher doses might work. Your aim should be to titrate up to the point of diminishing returns and then stabilize there or slightly pull back to the dose that gave the most recent very large benefit. Starting with the “flush,” going back to baseline, and then engaging in a slow upward titration allows you to more precisely define the dose you need without being fooled by it taking months to fix a deficiency.
The indications in two of these papers that variants in the hypoxia response could be creating blocks in the respiratory chain raise the possibility that some of the patients who never improved on riboflavin and sertraline withdrawal may have needed to fix their respiratory chain.
My Mitome test uses an interpretive algorithm that I invented that detects these variants of the hypoxia response using pattern analysis of the respiratory chain enzyme activity and generates a protocol to address each variant. This test does not diagnose or treat disease, but it can help with biochemical optimization where such bottlenecks in energy production are suspected.
We have now seen that both SSRI use and SSRI withdrawal can cause mitochondrial dysfunction, with poorly understood genetic idiosyncrasies determining which of these outcomes any given person lands on.
Playing with SSRIs is playing with fire.
The risk is you set your mitochondria on fire.
In future installments in this series, we will look at how to handle SSRI-associated mitochondrial dysfunction and at vastly superior approaches to mental health that can avoid these catastrophic results altogether.
Typo near beginning 250,0000.
But how can we fix this? How can someone heal or have better quality of life if they are effected?