

I hope all of those who celebrated had a wonderful Thanksgiving! Giving thanks is at least as healing as a healthy diet! For those of you who celebrated it just to eat a lot or for entirely other reasons, I hope you also had a wonderful holiday, and for those of you who did not celebrate anything at all, I hope you had a wonderful weekend.
In my last choline post, I presented my hypothesis that a suboptimal intake of choline, found most abundantly in egg yolks and organ meats like liver, is the preeminent cause of fatty liver disease. This disease may afflict 70-100 million Americans. If you missed it, you can find the post here:
After reading the research reviewed in that post, I'm quite convinced that the development of fatty liver is pretty simple: anything that increases the amount of energy that the liver needs to process forms the first part; anything that impairs the ability of the liver to export that energy forms the second part. High intakes of total calories, sugar, fat, or alcohol can all contribute to fatty liver in animals. But the king of all nutrients needed to export that energy as fat, choline, protects against the disease in all of these animal models.
Of course, a high intake of PUFA contributes to the inflammatory component, and when combined with other toxic factors may also contribute to the fatty component by preventing the liver from exporting its fat.
Does choline play the same role in fatty liver disease in humans? There are a few reasons to think it does. The most convincing of these is that it's been demonstrated in folks getting fed intravenously. The most interesting of these is the wide prevalence of a defect in the gene that allows us to synthesize phosphatidylcholine from the amino acid methionine. Its prevalence is high in the general population but almost complete among those with fatty liver.
The basic mechanisms of fatty liver in humans are the same as they are in animals. Humans with fatty liver disease have increased lipogenesis (synthesis of fat from carbohydrate) (1), increased circulation of fat released from adipose tissue (2), and decreased secretion of fat from the liver (3).
Although folks with fatty liver release more fatty acids from the fat tissue, the studies cited above show that their livers don't actually synthesize more fat from these fatty acids than the livers of healthy folks do. So those fatty acids may contribute to “lipotoxicity” — and in fact the creation of triglycerides in the liver is in part probably an effort to protect against the toxicity of excess free fatty acids — but it seems that the synthesis of fat from carbohydrate is more important than the release of fat from adipose stores.
The increased synthesis of fat almost certainly means that the burning of fat is decreased, because these two phenomena are metabolically linked to each other. Animal experiments using specific enzyme inhibitors suggest that both need to occur simultaneously to get significant increases in liver fat (4), if you want to induce it without any changes in the diet.
It was until quite recently thought that choline is not an essential nutrient in humans because choline can be synthesized from methionine. In 2001, however, a very small randomized, placebo-controlled trial conducted in 15 patients who developed fatty liver while receiving intravenous total parenteral nutrition (TPN) showed that the addition of two grams of choline chloride to the formula could completely resolve the fatty liver (5).
There are a number of other nutrients that reduce our requirement for choline in addition to methionine. One is betaine, which occurs most abundantly in wheat, spinach, and beets. Folate and vitamin B6 are also important. However, betaine, folate and B6 simply spare choline. We can actually use methionine to methylate a phospholipid called phosphatidylethanolamine to phosphatidylcholine, the principal choline-containing phospholipid.
There's just one problem: doing so requires an enzyme called phosphatidylethanolamine-N-methyltransferase (PEMT). The presence of at least one defective copy of this gene is extremely prevalent in the population, and it dramatically raises our chances of developing fatty liver on a low-choline diet.
Here's what one case-control study found when they compared the prevalence of the defective gene among people who had fatty liver and those who didn't (6):
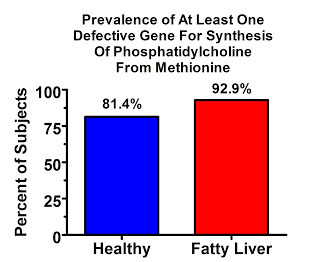
The vast majority of people in this study had at least one defective copy of the gene, but almost everyone who had fatty liver had at least one copy. The difference between the two groups doesn't seem huge, but if we spin the tables and look at who didn't have any copies of the defective gene, which is just another way of looking at the same data, the difference looks pretty big:
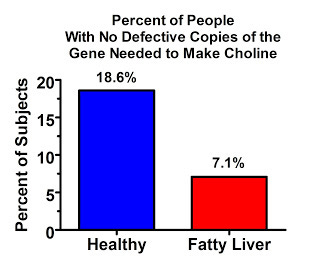
And here's the prevalence of people with two copies of the defective gene:
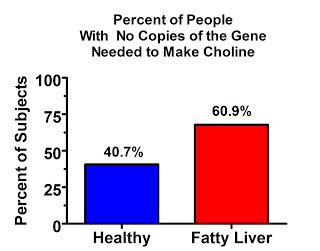
Of course “defective” is a value judgment. Apparently it's “good enough” to survive at a high prevalence. But I'll call it “defective” for now for the sake of simplicity.
One recent study suggests that people with defective copies of this gene are much more vulnerable to developing fatty liver on a low-choline diet.
The study is built on the findings of a previous study published a few years ago, so we'll discuss that one first. In the first study (7), the investigators fed 57 healthy men and women a diet containing roughly the RDA for choline (550 mg choline for every 70 kg bodyweight) for ten days. Then they switched them to a choline-deficient diet for 42 days. The diet was adequate (meeting or exceeding the RDA) for the other essential nutrients but contained less than 50 mg choline for every 70 kg bodyweight. The investigators then increased their choline intake stepwise, replacing 25% of it for ten days, then 50% for another ten days, then 75% for ten days, and then 100% for the final ten days.
38% of young women, 40% of men, and 80% of postmenopausal women developed fatty liver. 81% of these people resolved their fatty liver at some point during the repletion phase, while the others recovered only after returning to a normal diet.
This study is not terribly convincing. It is essentially an observational study because there was no control group, and rather than giving different people different doses of choline during the repletion period, they all got the same dose at once, increased over time. The results could be explained in numerous alternative ways. For example, the low-choline diet could have had some noxious factor in it that the subjects adversely reacted to at first, but to which most of them adapted over time.
Nevertheless, the study is an interesting pilot study, and it suggests that there may be very wide variability in the choline requirement, and that it may be much higher than the RDA in some people. And these people were healthy. As indicated by the genetic data above, people with fatty liver disease — possibly a third of the population — might have even higher needs for choline. And of course, the whole point of my last choline post was that the choline requirement is affected dramatically by what else we eat, not just our genes.
The second study (8) was designed similarly, but they did not monitor variation in the amount of choline needed for repletion. In this study, they divided postmenopausal women into those who had defective copies of the PEMT gene and those who did not, and they randomly allocated half of them to estrogen therapy. After all, the fact that twice as many postmenopausal women developed fatty liver in the previous study compared to younger women suggests that estrogen might reduce the choline requirement.
The results are pretty remarkable. The following results are for “organ dysfunction,” which included fatty liver, elevated liver enzymes, or signs of muscle dysfunction, but 90% of the women who developed “organ dysfunction” were diagnosed on the basis of fatty liver. The PEMT gene had a huge influence on who developed fatty liver:
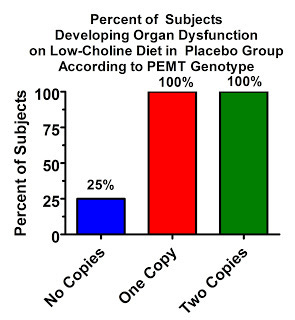
In the placebo group, only 25% of women with no defects in the PEMT gene developed “organ dysfunction” on the low-choline diet, but a whopping all of them developed organ dysfunction, virtually all of which was fatty liver, if they had at least one copy of the defective gene!
The estrogen had a remarkable protective effect. Two copies of the defective gene was very rare in this population (since the inclusion criteria required the participants be healthy), and no one was represented in the estrogen group. Nevertheless, even having one copy of the defective gene has a remarkable effect, as we'd expect based on the above graph:
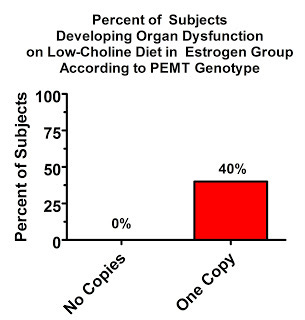
Quite an interaction! The combination of estrogen and two healthy copies of the PEMT gene abolishes the risk of choline-deficiency associated fatty liver, at least in the short course of a month, on an otherwise adequate diet. But it seems those with two of the “good” copies are in the minority.
The authors made the following remark about the effect of estrogen:
It makes sense, from an evolutionary perspective, that young women can supply more of their choline requirement from endogenous biosynthesis because pregnancy and lactation are times when the demand for choline is especially high, particularly to support the developing nervous system.
That's a rather odd comment considering the paper made no attempt to unravel the gentic mechanisms by which estrogen is increased during youth or by which estrogen decreases the choline requirement, but it's an interesting tangent. Certainly, it is biologically more important to provide choline to the brain of a developing fetus and infant than it is to make enough to get the fat out of your liver.
On the other hand, men were a lot better off than postmenopausal women, and although it's possible for men to lactate, it's very rare.
Evolutionarily, it makes a lot of sense for men not to lactate, because… ah well, nevermind. Maybe in another post.
Neither of these studies provide conclusive evidence that suboptimal intakes of choline cause fatty liver in humans, but together the totality of these data make a strong case for testing the hypothesis. It certainly seems that most of us are vulnerable to needing choline, rather than just methionine and the other nutrients that spare it. The TPN study provides proof of principle that choline does indeed reverse fatty liver in humans. And the universality of the protective effect of choline in diet-induced animal models of fatty liver certainly puts choline at center stage.
PEMT is not the only gene that affects the choline requirement. I'll be writing much more about the choline requirement and how to obtain it in a future post, and that will cover the effects of other genes as well.
Lets raise another glass to King Choline! Long live liver and eggs!
Read more about Chris Masterjohn, PhD, here.